|
# |
#
# |
#
 #
# Photograph by Martin Oeggerli, Micronaut, Supported by School of Life Sciences.
# # How do we make sense out of them?
#
# 
# # How do we make sense out of them?
#
# 
# # What is $\beta$-diversity
#
# - Comparison of two individual communities to determine how similar they are.
#
#
#
# Photograph by Martin Oeggerli, Micronaut, Supported by School of Life Sciences.
# # How do we make sense out of them?
#
# 
# # How do we make sense out of them?
#
# 
# # What is $\beta$-diversity
#
# - Comparison of two individual communities to determine how similar they are.
#
#  # # What is $\beta$-diversity
#
# - Comparison of two individual communities to determine how similar they are.
#
#
# # What is $\beta$-diversity
#
# - Comparison of two individual communities to determine how similar they are.
#
# 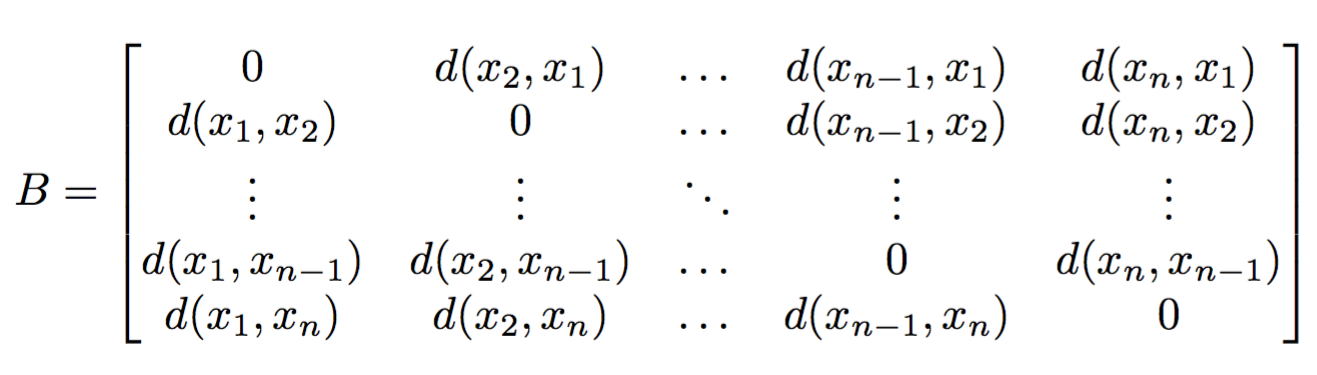 # # What is $\beta$-diversity
#
# - Comparison of two individual samples to determine how similar they are.
# - Elucidate patterns.
#
# 
#
#
# # What is $\beta$-diversity
#
# - Comparison of two individual samples to determine how similar they are.
# - Elucidate patterns.
#
# 
#
# # Bacterial community variation in human body habitats across space and time. # Costello EK et al. 2009. #
# # What is $\beta$-diversity # # - Comparison of two individual samples to determine how similar they are. # - Elucidate patterns. # #  # ## Bacterial community variation in human body habitats across space and time. # Costello EK et al. 2009. #
# # What is $\beta$-diversity # # - Comparison of two individual samples to determine how similar they are. # - Elucidate patterns. # #  # ## Bacterial community variation in human body habitats across space and time. # Costello EK et al. 2009. #
# In[4]: from skbio import OrdinationResults coordinates = OrdinationResults.read('costello/unweighted_unifrac_pc.txt') metadata = load_mf('costello/mapping-file.txt') # In[6]: coordinates # # In[7]: from emperor import Emperor # In[8]: Emperor(coordinates, metadata, remote=False) # # # # Outline # # - ~~Background (why $\beta$-diversity).~~ # # - **What is Emperor.** # # - How can we use Emperor. # # - Analyzing a use case. # # What is Emperor? # # - A Python 2/3 package that powers a JavaScript UI. # - https://github.com/biocore/emperor # # - Originated in the context of Quantitative Insights Into Microbial Ecology http://2.qiime.org # - Scatter plot viewer. # # - Visualize # - Interact # - Share # # Other applications # - Qiita # - https://qiita.ucsd.edu #  # - American Gut (participant's results). # - http://americangut.org/ # # - Illumina's BaseSpace processing results for the QIIME app.
# - Metabolomic analysis, IPython notebook ...
# ... kinda
# # Kinda?
#
# - https://github.com/jupyter/nbviewer/issues/316
#
#
# - Illumina's BaseSpace processing results for the QIIME app.
# - Metabolomic analysis, IPython notebook ...
# ... kinda
# # Kinda?
#
# - https://github.com/jupyter/nbviewer/issues/316
#
#  # # Nowadays
# - Python API.
# - Python 2 and 3.
# - Integration with scikit-bio.
# - Jupyter integration.
# - Pandas 🐼 Integration.
# - JavaScript API.
# - Command Line Interface (powered by QIIME 2).
# # Outline
#
# - ~~Background (why $\beta$-diversity).~~
#
# - ~~What is Emperor.~~
#
# - **How can we use Emperor**.
#
# - Analyzing a use case.
# # Python
# - 1 main class `Emperor`, depends on `scikit-bio` and `pandas`.
#
# - Format Python data into JSON and display it using JavaScript.
# In[9]:
from emperor import Emperor
get_ipython().run_line_magic('pinfo', 'Emperor')
#
# # http://emperor.microbio.me/uno/
#
# 
# # http://emperor.microbio.me/uno/
#
# 
# # http://emperor.microbio.me/uno/
#
# 
# # 🐼s integration - experimental
# In[10]:
import pandas as pd
# In[11]:
df = pd.read_csv('./tips.csv')
# In[12]:
df
#
# # 🐼s integration - experimental
# - Coordinates are inferred from the numerical data, see additional `x`, `y` and `z` parameters.
# In[13]:
from emperor import scatterplot
scatterplot(df, remote=False)
#
# # Outline
#
# - ~~Background (why $\beta$-diversity).~~
#
# - ~~What is Emperor.~~
#
# - **How can we use Emperor.**
#
# - Analyzing a use case.
# # Emperor and Jupyter 📓
#
# - nbviewer
# See the examples folder in our repo: https://github.com/biocore/emperor/tree/new-api/examples
#
# - Jupyter notebook
#
# - Standalone HTML plot
# - Generate a standalone HTML file with the needed resources.
#
# # JavaScript
# - Isn't this Sci**Py**.
# SciJS?
# - Thoroughly unit tested.
# - Public API ready to be used:
#
# http://emperor.microbio.me/uno/build/jsdoc/index.html
# # JavaScript
# 
# # JavaScript
# - Integration with SAGE2:
#
# http://sage2.sagecommons.org/
#
# # JavaScript
# - Integration with SAGE2:
#
# http://sage2.sagecommons.org/
#
# 
# # QIIME 2 integration
# - 1.5 hours to implement.
# - In less than 80 lines of code we got a CLI, GUI and provenance tracking.
# - Consider QIIME 2 as a gateway to an expanded user base.
# - CLI Provided through QIIME 2
#
# https://github.com/qiime2/qiime2
#
# https://github.com/qiime2/q2-emperor/
#
# ```bash
# qiime emperor plot --help
# ```
# # Outline
#
# - ~~Background (why $\beta$-diversity).~~
#
# - ~~What is Emperor.~~
#
# - ~~How can we use Emperor.~~
#
# - **Analyzing a use case.**
# # Use case
# - Let's leverage the technology we have available.
# - Create a small interface to:
#
# - Subsample.
# - Compute a distance matrix.
# * Use all the cores in our machine.
# - Visualize.
#
# - This was kinda possible through E-vident https://github.com/biocore/evident
# In[14]:
# biocore
from emperor.qiime_backports.parse import parse_mapping_file
from emperor import Emperor, nbinstall
nbinstall()
from skbio.stats.ordination import pcoa
from skbio.diversity import beta_diversity
from skbio import TreeNode
from skbio.io.util import open_file
from biom import load_table
from biom.util import biom_open
import qiime_default_reference
# pydata/scipy
import pandas as pd
import numpy as np
from scipy.spatial.distance import braycurtis, canberra
from ipywidgets import interact
from sklearn.metrics import pairwise_distances
from functools import partial
# In[15]:
import warnings
# don't try this at home
warnings.filterwarnings(action='ignore', category=Warning)
# -1 means all the processors available
pw_dists = partial(pairwise_distances, n_jobs=-1)
def load_mf(fn):
with open_file(fn) as f:
mapping_data, header, _ = parse_mapping_file(f)
_mapping_file = pd.DataFrame(mapping_data, columns=header)
_mapping_file.set_index('SampleID', inplace=True)
return _mapping_file
# # Load the data (table and tree)
# In[16]:
mf = load_mf('keyboard/mapping-file.txt')
bt = load_table('keyboard/otu-table.biom')
# In[17]:
tree = TreeNode.read(qiime_default_reference.get_reference_tree())
for n in tree.traverse():
if n.length is None:
n.length = 0
# # Interaction function
# In[18]:
def evident(n, metric):
rarefied = bt.subsample(n)
data = np.array([rarefied.data(i) for i in rarefied.ids()], dtype='int64')
# phylogenetic
if metric in ['unweighted_unifrac', 'weighted_unifrac']:
res = pcoa(beta_diversity(metric, data, rarefied.ids(),
otu_ids=rarefied.ids('observation'),
tree=tree, pairwise_func=pw_dists))
# non-phylogenetic
else:
res = pcoa(beta_diversity(metric, data, rarefied.ids(),
pairwise_func=pw_dists))
return Emperor(res, mf, remote=True)
# In[19]:
interact(evident, n=(200, 2000, 50),
metric=['unweighted_unifrac', 'weighted_unifrac', 'braycurtis', 'euclidean'],
__manual=True)
#
# # Summarizing
# Ready to be used!
# In[20]:
print(b'\xF0\x9F\x91\x8D'.decode('utf-8'))
# ## version 1.0 $\beta$ is out!
# ```bash
# pip install jupyter
# pip install emperor --pre
#
#
# conda install -c biocore emperor jupyter
# ```
# # Acknowledments
# - Thanks to all our users (**cited 99 times since 2013**)
# In[ ]:
from knightlab.members import current
from knightlab.members import past
from caporasolab.members import current as c_current
__credits__ = ['Antonio Gonzalez', 'Joshua Shorenstein', 'Jamie Morton',
'Jose Navas', 'Rob Knight'] + current + past + c_current
# In[21]:
one_more_thing()
#
# # Nowadays
# - Python API.
# - Python 2 and 3.
# - Integration with scikit-bio.
# - Jupyter integration.
# - Pandas 🐼 Integration.
# - JavaScript API.
# - Command Line Interface (powered by QIIME 2).
# # Outline
#
# - ~~Background (why $\beta$-diversity).~~
#
# - ~~What is Emperor.~~
#
# - **How can we use Emperor**.
#
# - Analyzing a use case.
# # Python
# - 1 main class `Emperor`, depends on `scikit-bio` and `pandas`.
#
# - Format Python data into JSON and display it using JavaScript.
# In[9]:
from emperor import Emperor
get_ipython().run_line_magic('pinfo', 'Emperor')
#
# # http://emperor.microbio.me/uno/
#
# 
# # http://emperor.microbio.me/uno/
#
# 
# # http://emperor.microbio.me/uno/
#
# 
# # 🐼s integration - experimental
# In[10]:
import pandas as pd
# In[11]:
df = pd.read_csv('./tips.csv')
# In[12]:
df
#
# # 🐼s integration - experimental
# - Coordinates are inferred from the numerical data, see additional `x`, `y` and `z` parameters.
# In[13]:
from emperor import scatterplot
scatterplot(df, remote=False)
#
# # Outline
#
# - ~~Background (why $\beta$-diversity).~~
#
# - ~~What is Emperor.~~
#
# - **How can we use Emperor.**
#
# - Analyzing a use case.
# # Emperor and Jupyter 📓
#
# - nbviewer
# See the examples folder in our repo: https://github.com/biocore/emperor/tree/new-api/examples
#
# - Jupyter notebook
#
# - Standalone HTML plot
# - Generate a standalone HTML file with the needed resources.
#
# # JavaScript
# - Isn't this Sci**Py**.
# SciJS?
# - Thoroughly unit tested.
# - Public API ready to be used:
#
# http://emperor.microbio.me/uno/build/jsdoc/index.html
# # JavaScript
# 
# # JavaScript
# - Integration with SAGE2:
#
# http://sage2.sagecommons.org/
#
# # JavaScript
# - Integration with SAGE2:
#
# http://sage2.sagecommons.org/
#
# 
# # QIIME 2 integration
# - 1.5 hours to implement.
# - In less than 80 lines of code we got a CLI, GUI and provenance tracking.
# - Consider QIIME 2 as a gateway to an expanded user base.
# - CLI Provided through QIIME 2
#
# https://github.com/qiime2/qiime2
#
# https://github.com/qiime2/q2-emperor/
#
# ```bash
# qiime emperor plot --help
# ```
# # Outline
#
# - ~~Background (why $\beta$-diversity).~~
#
# - ~~What is Emperor.~~
#
# - ~~How can we use Emperor.~~
#
# - **Analyzing a use case.**
# # Use case
# - Let's leverage the technology we have available.
# - Create a small interface to:
#
# - Subsample.
# - Compute a distance matrix.
# * Use all the cores in our machine.
# - Visualize.
#
# - This was kinda possible through E-vident https://github.com/biocore/evident
# In[14]:
# biocore
from emperor.qiime_backports.parse import parse_mapping_file
from emperor import Emperor, nbinstall
nbinstall()
from skbio.stats.ordination import pcoa
from skbio.diversity import beta_diversity
from skbio import TreeNode
from skbio.io.util import open_file
from biom import load_table
from biom.util import biom_open
import qiime_default_reference
# pydata/scipy
import pandas as pd
import numpy as np
from scipy.spatial.distance import braycurtis, canberra
from ipywidgets import interact
from sklearn.metrics import pairwise_distances
from functools import partial
# In[15]:
import warnings
# don't try this at home
warnings.filterwarnings(action='ignore', category=Warning)
# -1 means all the processors available
pw_dists = partial(pairwise_distances, n_jobs=-1)
def load_mf(fn):
with open_file(fn) as f:
mapping_data, header, _ = parse_mapping_file(f)
_mapping_file = pd.DataFrame(mapping_data, columns=header)
_mapping_file.set_index('SampleID', inplace=True)
return _mapping_file
# # Load the data (table and tree)
# In[16]:
mf = load_mf('keyboard/mapping-file.txt')
bt = load_table('keyboard/otu-table.biom')
# In[17]:
tree = TreeNode.read(qiime_default_reference.get_reference_tree())
for n in tree.traverse():
if n.length is None:
n.length = 0
# # Interaction function
# In[18]:
def evident(n, metric):
rarefied = bt.subsample(n)
data = np.array([rarefied.data(i) for i in rarefied.ids()], dtype='int64')
# phylogenetic
if metric in ['unweighted_unifrac', 'weighted_unifrac']:
res = pcoa(beta_diversity(metric, data, rarefied.ids(),
otu_ids=rarefied.ids('observation'),
tree=tree, pairwise_func=pw_dists))
# non-phylogenetic
else:
res = pcoa(beta_diversity(metric, data, rarefied.ids(),
pairwise_func=pw_dists))
return Emperor(res, mf, remote=True)
# In[19]:
interact(evident, n=(200, 2000, 50),
metric=['unweighted_unifrac', 'weighted_unifrac', 'braycurtis', 'euclidean'],
__manual=True)
#
# # Summarizing
# Ready to be used!
# In[20]:
print(b'\xF0\x9F\x91\x8D'.decode('utf-8'))
# ## version 1.0 $\beta$ is out!
# ```bash
# pip install jupyter
# pip install emperor --pre
#
#
# conda install -c biocore emperor jupyter
# ```
# # Acknowledments
# - Thanks to all our users (**cited 99 times since 2013**)
# In[ ]:
from knightlab.members import current
from knightlab.members import past
from caporasolab.members import current as c_current
__credits__ = ['Antonio Gonzalez', 'Joshua Shorenstein', 'Jamie Morton',
'Jose Navas', 'Rob Knight'] + current + past + c_current
# In[21]:
one_more_thing()
# |
# |
#
# |
#