Getting meaning from scientific articles¶
1- Who I am
2- Goal of this project
3- My plan
4- First iteration
5- Improvements
6- Second iteration
7- Discussion
Getting meaning from scientific articles¶
1- Who I am
2- Goal of this project
3- My plan
4- First iteration
5- Improvements
6- Second iteration
7- Discussion
Who I am¶
Background in biomedical research
Learned Python at an intensive course
Now developer, data wrangler, hardware tinkerer
=> What would I have done had I known how to code back in my lab days?
Getting meaning from scientific articles¶
1- Who I am
2- Goal of this project
3- My plan
4- First iteration
5- Improvements
6- Second iteration
7- Discussion
Goal of the project¶
Problem: Bibliography is an important activity that takes time off research work.

-> Could the bibliography process be less time-consuming?
Idea: What if it was possible to classify a new article in one of those sub-categories
Getting meaning from scientific articles¶
1- Who I am
2- Goal of this project
3- My plan
4- First iteration
5- Improvements
6- Second iteration
7- Discussion
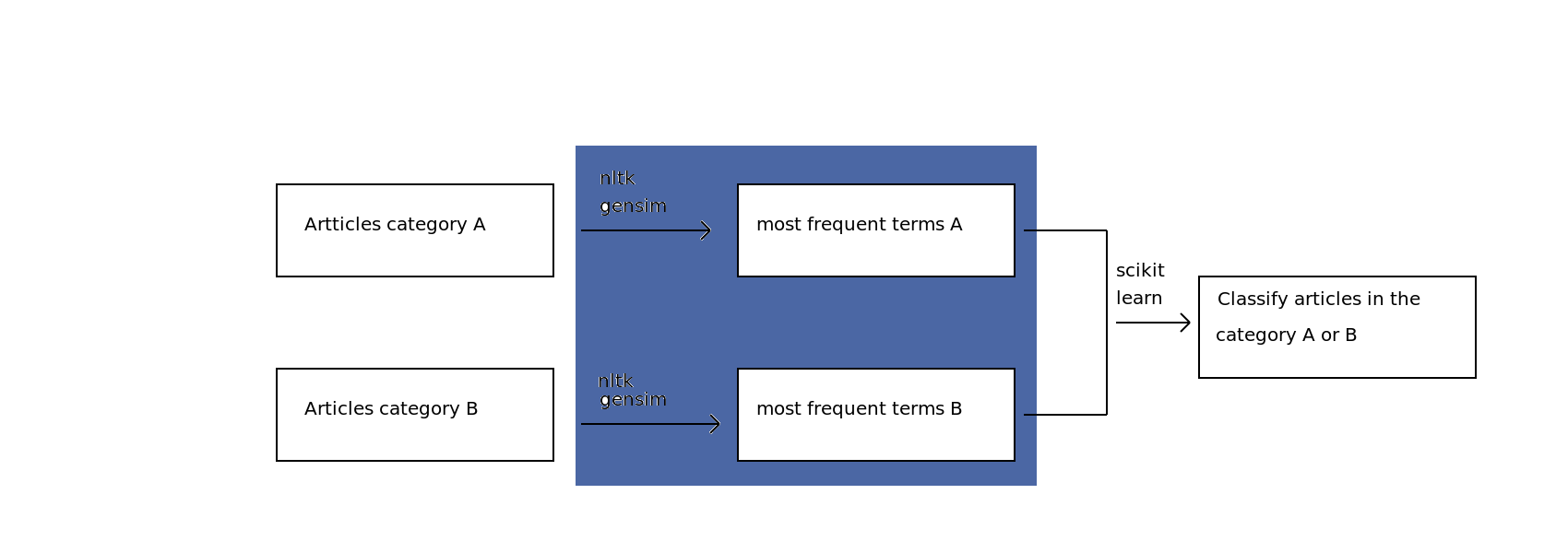
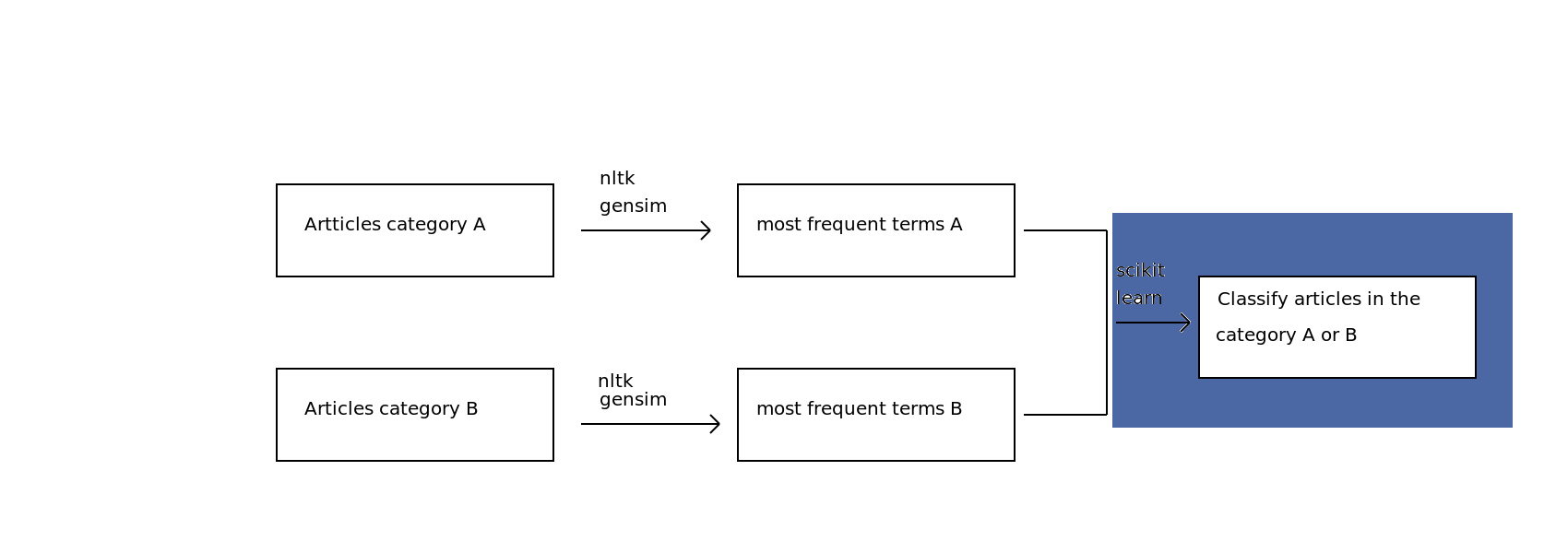
My plan¶
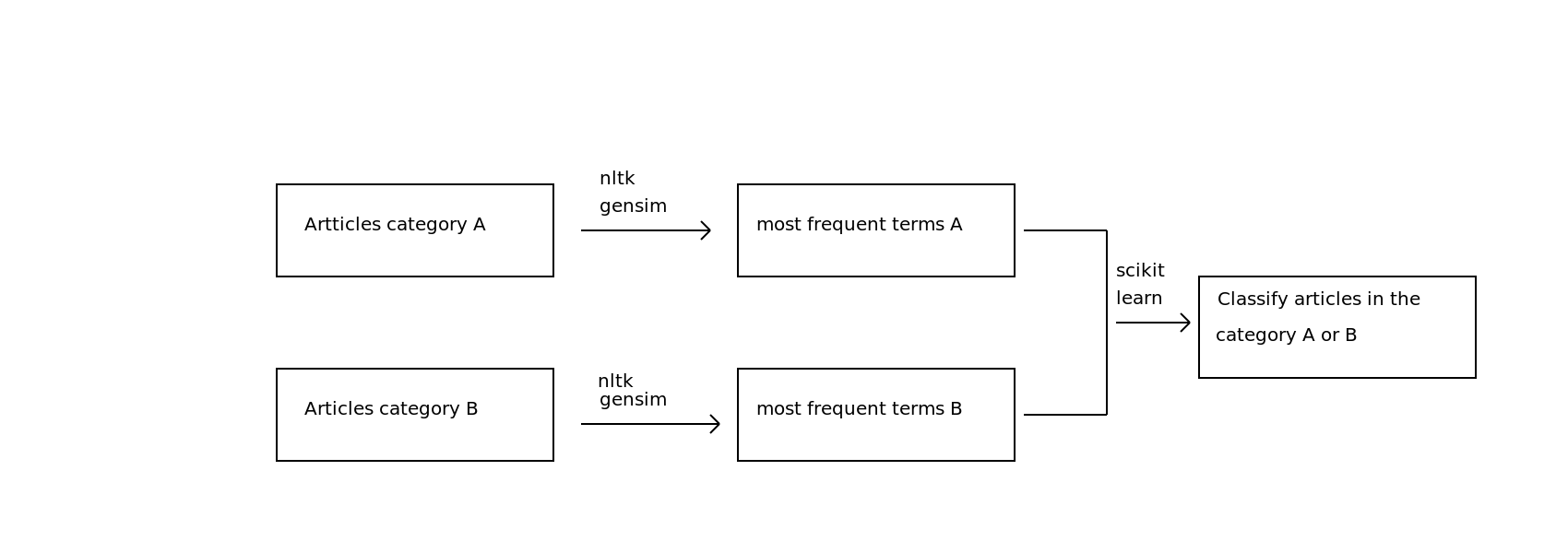
Getting meaning from scientific articles¶
1- Who I am
2- Goal of this project
3- My plan
4- First iteration
5- Improvements
6- Second iteration
7- Discussion

First iteration¶
STEP 1: Gathering scientific articles
1 - Yay for Open Access research articles!
2 - Hmmm, where's the API so I can upload hundreds of them?
3 - My solution: eLifeScience articles on Github (http://github.com/elifesciences/elife-articles)
4 - Parsing XML files using Beautiful Soup -> Group A: "Neuroscience" - Group B: "Cell biology"
First iteration¶
STEP 2: Retrieve the most frequent terms
-> Use Nltk and Gensim libraries to implement the TF-IDF (Term frequency–inverse document frequency) model
->Write functions to be executed on a collection of text files (converted from XML)
# %load get_topics.py
# now renamed scripts/lda_topics.py
#!/home/eleonore/virtenvs/nltk-gensim-skl/bin/python2.7
from nltk.stem.wordnet import WordNetLemmatizer
from gensim import models
from gensim.corpora import Dictionary
from os import listdir
from os.path import isfile, join
import string, re, codecs, time, json
## Global variables
stop_words = ['a', 'also', 'an', 'and', 'are', 'as', 'at', 'be', 'by', 'but', 'for',
'from', 'has', 'he', 'if', 'in', 'is', 'it', 'its', 'it\'s', 'not',
'of', 'on', 'our', 'than', 'that', 'the', 'therefore', 'to', 'was',
'were', 'will', 'with', 'may', 'need', 'have', 'been', 'their', 'this',
'these', 'which', 'do', 'did', 'red', 'blue', 'green', 'bar', 'chart',
'arrowhead', 'arrow', 'vice', 'versa']
spe_char = {u'β': 'beta', u'α': 'alpha', u'µm': 'micron'}
## Functions to break up the process:
def parse_text(text_file):
"Gets a text file outputs a list of strings."
with codecs.open(text_file, mode='r', encoding='utf-8') as f:
read = f.read()
r = [read.replace(unicode(i), spe_char.get(i)) for i in read if i in spe_char.keys()] or [read]
text = [line for line in r[0].strip().split('. ') if line != '']
return text
def get_tokens(text_parsed):
"Gets a text and retrieves tokens."
# Tokenisation
texts = [t.lower().replace('\n', ' ').split(' ') for t in text_parsed]
# Remove punctuation and stop words
tokens = [[filter(lambda x:x not in string.punctuation, i)
for i in txt if i != '' and i not in stop_words] for txt in texts]
tokens_cleaned = [[i for i in txt if len(i) > 2 and not i.isdigit()] for txt in tokens]
return tokens_cleaned
def lemmatize_tokens(tokens):
"Gets tokens and retrieves lemmatised tokens."
# Lemmatisation using nltk lemmatiser
lmtzr = WordNetLemmatizer()
lemma = [[lmtzr.lemmatize(word) for word in data] for data in tokens]
return lemma
def bag_of_words(lemma):
"Takes in lemmatised words and returns a bow."
## Create bag of words from dictionnary
dictionary = Dictionary(lemma)
dictionary.save('../dicts/bow-text.dict')
## Term frequency–inverse document frequency (TF-IDF)
bow = [dictionary.doc2bow(l) for l in lemma]
return bow
def tfidf_and_lsi(lemma, bow):
"Gets a bow and returns topics."
dictionary = Dictionary(lemma)
# Transform the count representation into the Tfidf space
tfidf = models.TfidfModel(bow)
corpus_tfidf = tfidf[bow]
## Build the LSI model
lsi = models.LsiModel(corpus_tfidf, id2word=dictionary, num_topics=6)
corpus_lsi = lsi[corpus_tfidf]
list_topics = []
for i in range(lsi.num_topics):
list_topics.extend(lsi.show_topic(i))
list_topics.sort(key=lambda tup: tup[0], reverse=True)
topics = [i[1] for i in list_topics[:10]]
return topics
## Function to retrieve topics using nltk
def get_topics(text_file):
txt = parse_text(text_file)
tokens = get_tokens(txt)
lemma = lemmatize_tokens(tokens)
bow = bag_of_words(lemma)
return tfidf_and_lsi(lemma, bow)
## Get all text articles from a path and retrieve topics:
def list_all_articles(path):
articles = [f for f in listdir(path) if isfile(join(path, f))] or []
print "There are %d articles in %s" % (len(articles), path)
return {"path": path, "articles": articles}
# Write the topics to a json file:
#{"Neuroscience": {"pub_id1":[topics_file1], "pub_id2":[topics_file2]...},
# "Cell biology": {[], []...}}
def get_articles_topics(path, filename):
"Store the topics in a json object and dump to a file."
all_topics = {}
#Get the directories in a path
dirs = [d for d in listdir(path) if not isfile(join(path, d))]
#For each dir and for each file in a dir
for d in dirs:
all_topics[d] = {}
txt_files = [f for f in listdir(path+d) if isfile(join(path+d, f))]
print txt_files
for f in txt_files:
all_topics[d][f[:-4]] = get_topics(path+d+'/'+f)
with open(filename, 'w') as f:
json.dump(all_topics, f)
return all_topics
if __name__ == "__main__":
neuro_articles = list_all_articles("../articles/Neuroscience/")
cellbiol_articles = list_all_articles("../articles/Cell biology/")
print get_topics(neuro_articles.get("path") + neuro_articles.get("articles")[0])
#print get_articles_topics("../articles/", "../json/filenames_topics.json")
There are 172 articles in articles/Neuroscience/ There are 67 articles in articles/Cell biology/ [u'scale', u'colocalization', u'vglut1', u'pearsons', u'coefficient', u'panel', u'red', u'colocalization', u'entire', u'hippocampal']
First iteration¶
STEP 3: Build a classifier to predict the subcategory of an article
-> Exploring the data with Numpy and Scikit-learn
import json
import matplotlib.pyplot as plt
import numpy as np
import scipy as sp
from scipy import stats
import sklearn
%matplotlib inline
plt.style.use('ggplot')
with open("../json/filenames_topics.json", 'r') as f:
topics_articles = json.load(f)
print topics_articles['Neuroscience']['elife05116']
print topics_articles['Cell biology']['elife02923']
[u'scale', u'detected', u'inset', u'could', u'scale', u'rnai', u'antiphosphoret', u'development', u'control', u'receptor'] [u'allele', u'marker', u'vastus', u'lateralis', u'staining', u'ryr1ag', u'old', u'arrow', u'1year', u'background']
# List of most frequent terms
header = ['article', 'subject']
for subject, articles in topics_articles.iteritems():
for pub_id, topics in articles.iteritems():
header.extend(topics)
print header[:15]
# Matrix representing the presence or absence of those terms in each article
top_data = []
for subject, articles in topics_articles.iteritems():
for pub_id, topics in articles.iteritems():
ct = []
ct.append(pub_id)
ct.append(subject)
tpcs = ['1' if h in topics else '0' for h in header[2:]]
ct.extend(tpcs)
top_data.append(ct)
print top_data[0][:15]
['article', 'subject', u'scale', u'detected', u'inset', u'could', u'scale', u'rnai', u'antiphosphoret', u'development', u'control', u'receptor', u'reduced', u'hand', u'reduced'] [u'elife05116', u'Neuroscience', '1', '1', '1', '1', '1', '1', '1', '1', '1', '1', '0', '0', '0']
# Use a numpy array (data structure used with Scikit-learn)
topics_data = np.array(top_data)
print topics_data
## Data matrix: column 3 to the end
X = topics_data[:, 2:2392].astype(int)
## Class vector: column 2
Y = topics_data[:, 1]
[[u'elife05116' u'Neuroscience' u'1' ..., u'0' u'0' u'0'] [u'elife02094' u'Neuroscience' u'0' ..., u'0' u'0' u'0'] [u'elife01206' u'Neuroscience' u'0' ..., u'0' u'0' u'0'] ..., [u'elife04810' u'Cell biology' u'0' ..., u'0' u'0' u'0'] [u'elife02678' u'Cell biology' u'0' ..., u'0' u'0' u'0'] [u'elife05697' u'Cell biology' u'1' ..., u'1' u'1' u'1']]
print "\nX dimension: ", X.shape
print "Y dimension: ", Y.shape
Yfreq = sp.stats.itemfreq(Y)
print Yfreq
cb_total = int(Yfreq[0][1])
n_total = int(Yfreq[1][1])
X dimension: (239, 2390) Y dimension: (239,) [[u'Cell biology' u'67'] [u'Neuroscience' u'172']]
freqs_cb, freqs_n = [], []
R, N = range(3, 15, 2), len(range(3, 15, 2))
for j in R:
cb = [int(i[1]) for i in topics_data[:, (1, j)] if i[0]=='Cell biology']
n = [int(i[1]) for i in topics_data[:, (1, j)] if i[0]=='Neuroscience']
cb_freq = sum(cb)*100./cb_total
n_freq = sum(n)*100./n_total
freqs_cb.append(cb_freq)
freqs_n.append(n_freq)
ind = np.arange(N)
width = 0.3
fig, ax = plt.subplots()
rects_cb = ax.bar(ind, freqs_cb, width, color='#A770BF')
rects_n = ax.bar(ind+width, freqs_n, width, color='#81ABD3')
ax.set_ylabel('Percentage of occurence')
ax.set_title('Occurence of frequent term in Cell biology vs Neuroscience')
ax.set_xticks(ind+width)
xlabels = [header[i] for i in R]
ax.set_xticklabels(xlabels)
ax.legend((rects_cb[0], rects_n[0]), ('Cell biology', 'Neuroscience'))
fig.subplots_adjust(right=2)

#x_index, y_index = 48, 350
x_index, y_index = 859, 179
classes = ['Cell biology', 'Neuroscience']
formatter = plt.FuncFormatter(lambda i, *args: classes[int(i)])
from sklearn import preprocessing
le = preprocessing.LabelEncoder()
le.fit(Y)
targets = le.transform(Y)
plt.scatter(topics_data[:, x_index], topics_data[:, y_index],
s=50, c=targets, alpha=0.5)
plt.colorbar(ticks=[0, 1], format=formatter)
plt.clim(-0.2, 1.2)
plt.xlabel(header[x_index])
plt.ylabel(header[y_index])
<matplotlib.text.Text at 0x7f2eef212a90>

How I executed my plan¶
STEP 3: Build a classifier to predict the subcategory of an article
-> Building a classifier
Naive Bayes classifier:
Simple classifier applying Bayes' theorem
Assumes independence of the features
Widely used in different application (text categorisation)
Doesn't require a very large dataset
## NAIVE BAYES classifier:
from sklearn.naive_bayes import BernoulliNB
from sklearn import metrics, preprocessing
from sklearn.cross_validation import train_test_split
## Encode labels ('Cell biology', 'Neuroscience') as 0 and 1.
le = preprocessing.LabelEncoder()
le.fit(Y)
y_transformed = le.transform(Y)
#print Y
#print y_transformed
## => 1: Neuroscience, 0: Cell biology
## Randomly split the data between training and testing:
X_train, X_test, Y_train, Y_test = train_test_split(X, y_transformed)
## Bernoulli NB classifier
nbmodel_train = BernoulliNB().fit(X_train, Y_train)
predicted_bnb = nbmodel_train.predict(X_test)
## Confusion matrix:
cm_bnb = metrics.confusion_matrix(Y_test, predicted_bnb)
print "True positive: ", cm_bnb[0][0]," - False negative: ", cm_bnb[0][1]
print "False positive: ", cm_bnb[1][0], " - True negative: ", cm_bnb[1][1]
True positive: 10 - False negative: 7 False positive: 3 - True negative: 40
## Bernoulli NB classifier
# Precision: fraction of retrieved instances that are relevant (TP / TP + FP)
# Recall: fraction of relevant instances that are retrieved
print metrics.classification_report(Y_test, predicted_bnb)
# Accuracy: overall correctness of the model
print "Accuracy: ", metrics.accuracy_score(Y_test, predicted_bnb)
## => Test with a high precision and recall for class 1 = Neuroscience
precision recall f1-score support
0 0.77 0.59 0.67 17
1 0.85 0.93 0.89 43
avg / total 0.83 0.83 0.83 60
Accuracy: 0.833333333333
K-nearest neighbor algorithm:
Predict the label of a new point from its neighbours
Simplest classifier in Machine Learning
Lazy learner: computationally demanding for large datasets
## K NEAREST NEIGHBOUR classifier
from sklearn import metrics
from sklearn import neighbors
X_train, X_test, Y_train, Y_test = train_test_split(X, y_transformed)
knn = neighbors.KNeighborsClassifier(n_neighbors=3).fit(X_train, Y_train)
predicted_knn = knn.predict(X_test)
cm_knn = metrics.confusion_matrix(Y_test, predicted_knn)
print "True positive: %d - False negative: %d" % (cm_knn[0][0], cm_knn[0][1])
print "False positive: %d - True negative: %d \n" % (cm_knn[1][0], cm_knn[1][1])
True positive: 8 - False negative: 10 False positive: 5 - True negative: 37
## K NEAREST NEIGHBOUR classifier
# Precision: fraction of retrieved instances that are relevant (TP / TP + FP)
# Recall: fraction of relevant instances that are retrieved
print metrics.classification_report(Y_test, predicted_knn)
# Accuracy: overall correctness of the model
print "Accuracy: ", metrics.accuracy_score(Y_test, predicted_knn)
## => Test with a high precision and recall for class 1 = Neuroscience
precision recall f1-score support
0 0.62 0.44 0.52 18
1 0.79 0.88 0.83 42
avg / total 0.74 0.75 0.74 60
Accuracy: 0.75
## Model validation: test several training/testing data
# K fold cross validation: iterator that randomise the sampling
from sklearn.cross_validation import cross_val_score
kf = range(2, 21)
knn = neighbors.KNeighborsClassifier(n_neighbors = 3)
nb = BernoulliNB()
means_knn, sds_knn, means_nb, sds_nb = [], [], [], []
for k in kf:
knn_scores = cross_val_score(knn, X_train, Y_train, cv=k)
nb_scores = cross_val_score(nb, X_train, Y_train, cv=k)
means_knn.append(knn_scores.mean())
sds_knn.append(knn_scores.std())
means_nb.append(nb_scores.mean())
sds_nb.append(nb_scores.std())
print means_knn
print sds_knn
[0.74307116104868909, 0.74854126146151712, 0.77028436539306111, 0.77063492063492067, 0.75300333704115685, 0.7373626373626373, 0.72686100131752307, 0.7321080478975216, 0.74248366013071898, 0.73689839572192506, 0.75823412698412695, 0.73710904480135242, 0.74731815803244372, 0.74654234654234641, 0.74300699300699302, 0.74581105169340467, 0.73591470258136926, 0.75804093567251463, 0.75569444444444445] [0.0097378277153558068, 0.0039294625239596681, 0.036849480657926544, 0.049525335751818091, 0.055005815837847118, 0.035175822459327585, 0.042045989963185822, 0.054404434025065819, 0.064550549673863492, 0.065473035936487706, 0.063142873477736575, 0.06091209095622617, 0.061086699560089427, 0.083182950324958621, 0.093804744232841353, 0.08805579810894619, 0.077954833834140022, 0.084796581962805637, 0.089237489180885435]
plt.plot(kf, means_knn, label="KNN 3 mean accuracy", color="purple")
plt.plot(kf, means_nb, label="NB mean accuracy", color="blue")
plt.legend(loc=2)
plt.ylim(0.6, 0.9)
plt.title("Accuracy means with increasing K-fold")
<matplotlib.text.Text at 0x7f2eeb885c10>

plt.plot(kf, sds_knn, label="KNN 3 sds accuracy", color="purple")
plt.plot(kf, sds_nb, label="NB sds accuracy", color="blue")
plt.legend(loc=2)
plt.ylim(0.01, 0.4)
plt.title("Accuracy standard deviation")
<matplotlib.text.Text at 0x7f2eeb7c2c50>

## 10 folds
X_train, X_test, Y_train, Y_test = train_test_split(X, y_transformed)
NB_train = BernoulliNB()
for i in range(10):
NB_train.fit(X_train, Y_train)
import get_topics as gt
topics_test1 = gt.get_topics("../articles/conrad2013_melanoma.txt")
print topics_test1
topics_test2 = gt.get_topics("../articles/panizzo2014_cerebral_ischaemia.txt")
print topics_test2
test1 = [1 if h in topics_test1 else 0 for h in header[2:]]
test2 = [1 if h in topics_test2 else 0 for h in header[2:]]
NB_test1 = NB_train.predict(test1)
print NB_test1
NB_test2 = NB_train.predict(test2)
print NB_test2
[u'mouse', u'pathway', u'regulator', u'integrating', u'potential', u'identified', u'b16', u'data', u'pathway', u'potential'] [u'over', u'week', u'postischaemia', u'hour', u'cerebral', u'evolution', u'hypointense', u'cerebral', u'ischaemia', u'ischaemia'] [1] [1]
Getting meaning from scientific articles¶
1- Who I am
2- Goal of this project
3- My plan
4- First iteration
5- Improvements
6- Second iteration
7- Discussion
Improvements¶
Use article abstracts only
Use Python 3 to avoid unicode issues
Use the Latent Dirichet Allocation model
Second iteration¶
Topic modeling
"A topic model is a type of statistical model for discovering the abstract "topics" that occur in a collection of documents."
- Latent Semantic Analysis (LSA)
-> Analyse relationships between a set of documents and the terms they contain by producing a set of concepts related to the documents and terms.
- Latent Derichelet Allocation (LDA)
-> Differs from LSA by assuming each document to be characterized by a mixture of various topics.
Second iteration¶
Latent Derichelet Allocation (LDA) with Gensim
from nltk.stem.wordnet import WordNetLemmatizer
from gensim import models
from gensim.corpora import Dictionary
from gensim.parsing.preprocessing import STOPWORDS
from os import listdir
from os.path import isfile, join
import string
import codecs
import time
import json
# Testing data: 2 Cell bio article abstracts
conrad2013 = """The inability of targeted BRAF inhibitors to produce long-lasting improvement in the clinical outcome of melanoma highlights a need to identify additional approaches to inhibit melanoma growth.
Recent studies have shown that activation of the Wnt/β-catenin pathway decreases tumor growth and cooperates with ERK/MAPK pathway inhibitors to promote apoptosis in melanoma.
Therefore, the identification of Wnt/β-catenin regulators may advance the development of new approaches to treat this disease.
In order to move towards this goal we performed a large scale small-interfering RNA (siRNA) screen for regulators of β-catenin activated reporter activity in human HT1080 fibrosarcoma cells.
Integrating large scale siRNA screen data with phosphoproteomic data and bioinformatics enrichment identified a protein, FAM129B, as a potential regulator of Wnt/β-catenin signaling.
Functionally, we demonstrated that siRNA-mediated knockdown of FAM129B in A375 and A2058 melanoma cell lines inhibits WNT3A-mediated activation of a β-catenin-responsive luciferase reporter and inhibits expression of the endogenous Wnt/β-catenin target gene, AXIN2.
We also demonstrate that FAM129B knockdown inhibits apoptosis in melanoma cells treated with WNT3A. These experiments support a role for FAM129B in linking Wnt/β-catenin signaling to apoptosis in melanoma.
The incidence of melanoma continues to rise across the U.S. at a rate faster than any other cancer. Malignant melanoma has a poor prognosis with a 5-year survival rate of only 15%3.
The recently approved therapeutic, vemurafenib, extends median patient survival by 7 months. This major advance raises expectations that even greater rates of survival might be attainable with combination therapies."""
huang2015 = """Mouse GnT1IP-L, and membrane-bound GnT1IP-S (MGAT4D) expressed in cultured cells inhibit MGAT1, the N-acetylglucosaminyltransferase that initiates the synthesis of hybrid and complex N-glycans.
However, it is not known where in the secretory pathway GnT1IP-L inhibits MGAT1, nor whether GnT1IP-L inhibits other N-glycan branching N-acetylglucosaminyltransferases of the medial Golgi. We show here that the luminal domain of GnT1IP-L contains its inhibitory activity.
Retention of GnT1IP-L in the endoplasmic reticulum (ER) via the N-terminal region of human invariant chain p33, with or without C-terminal KDEL, markedly reduced inhibitory activity.
Dynamic fluorescent resonance energy transfer (FRET) and bimolecular fluorescence complementation (BiFC) assays revealed homomeric interactions for GnT1IP-L in the ER, and heteromeric interactions with MGAT1 in the Golgi.
GnT1IP-L did not generate a FRET signal with MGAT2, MGAT3, MGAT4B or MGAT5 medial Golgi GlcNAc-tranferases.
GnT1IP/Mgat4d transcripts are expressed predominantly in spermatocytes and spermatids in mouse, and are reduced in men with impaired spermatogenesis."""
spe_char = {u'β': 'beta', u'α': 'alpha', u'µm': 'micron'}
def parse_text(text):
"Gets a text and outputs a list of strings."
doc = [text.replace(unicode(i), spe_char.get(i)) for i in text if i in spe_char.keys()] or [text]
return doc
conrad2013_parsed = parse_text(conrad2013)
huang2015_parsed = parse_text(huang2015)
print conrad2013_parsed
['The inability of targeted BRAF inhibitors to produce long-lasting improvement in the clinical outcome of melanoma highlights a need to identify additional approaches to inhibit melanoma growth. \n Recent studies have shown that activation of the Wnt/\xce\xb2-catenin pathway decreases tumor growth and cooperates with ERK/MAPK pathway inhibitors to promote apoptosis in melanoma. \n Therefore, the identification of Wnt/\xce\xb2-catenin regulators may advance the development of new approaches to treat this disease. \n In order to move towards this goal we performed a large scale small-interfering RNA (siRNA) screen for regulators of \xce\xb2-catenin activated reporter activity in human HT1080 fibrosarcoma cells. \n Integrating large scale siRNA screen data with phosphoproteomic data and bioinformatics enrichment identified a protein, FAM129B, as a potential regulator of Wnt/\xce\xb2-catenin signaling. \n Functionally, we demonstrated that siRNA-mediated knockdown of FAM129B in A375 and A2058 melanoma cell lines inhibits WNT3A-mediated activation of a \xce\xb2-catenin-responsive luciferase reporter and inhibits expression of the endogenous Wnt/\xce\xb2-catenin target gene, AXIN2. \n We also demonstrate that FAM129B knockdown inhibits apoptosis in melanoma cells treated with WNT3A. These experiments support a role for FAM129B in linking Wnt/\xce\xb2-catenin signaling to apoptosis in melanoma.\n The incidence of melanoma continues to rise across the U.S. at a rate faster than any other cancer. Malignant melanoma has a poor prognosis with a 5-year survival rate of only 15%3. \n The recently approved therapeutic, vemurafenib, extends median patient survival by 7 months. This major advance raises expectations that even greater rates of survival might be attainable with combination therapies.']
/usr/local/lib/python2.7/dist-packages/IPython/kernel/__main__.py:5: UnicodeWarning: Unicode equal comparison failed to convert both arguments to Unicode - interpreting them as being unequal
def get_tokens(text_parsed):
"Gets a text and retrieves tokens."
# Tokenisation
texts = text_parsed[0].lower().replace('\n', ' ').replace('/', ' ').replace('-', ' ').split(' ')
# Remove punctuation and stop words
tokens = [filter(lambda x:x not in string.punctuation, i)
for i in texts if i != '' and i not in STOPWORDS]
tokens_cleaned = [i for i in tokens if len(i) > 2 and not i.isdigit()]
return tokens_cleaned
conrad2013_tokens = get_tokens(conrad2013_parsed)
huang2015_tokens = get_tokens(huang2015_parsed)
print conrad2013_tokens
['inability', 'targeted', 'braf', 'inhibitors', 'produce', 'long', 'lasting', 'improvement', 'clinical', 'outcome', 'melanoma', 'highlights', 'need', 'identify', 'additional', 'approaches', 'inhibit', 'melanoma', 'growth', 'recent', 'studies', 'shown', 'activation', 'wnt', 'catenin', 'pathway', 'decreases', 'tumor', 'growth', 'cooperates', 'erk', 'mapk', 'pathway', 'inhibitors', 'promote', 'apoptosis', 'melanoma', 'therefore', 'identification', 'wnt', 'catenin', 'regulators', 'advance', 'development', 'new', 'approaches', 'treat', 'disease', 'order', 'goal', 'performed', 'large', 'scale', 'small', 'interfering', 'rna', 'sirna', 'screen', 'regulators', 'catenin', 'activated', 'reporter', 'activity', 'human', 'ht1080', 'fibrosarcoma', 'cells', 'integrating', 'large', 'scale', 'sirna', 'screen', 'data', 'phosphoproteomic', 'data', 'bioinformatics', 'enrichment', 'identified', 'protein', 'fam129b', 'potential', 'regulator', 'wnt', 'catenin', 'signaling', 'functionally', 'demonstrated', 'sirna', 'mediated', 'knockdown', 'fam129b', 'a375', 'a2058', 'melanoma', 'cell', 'lines', 'inhibits', 'wnt3a', 'mediated', 'activation', 'catenin', 'responsive', 'luciferase', 'reporter', 'inhibits', 'expression', 'endogenous', 'wnt', 'catenin', 'target', 'gene', 'axin2', 'demonstrate', 'fam129b', 'knockdown', 'inhibits', 'apoptosis', 'melanoma', 'cells', 'treated', 'wnt3a', 'experiments', 'support', 'role', 'fam129b', 'linking', 'wnt', 'catenin', 'signaling', 'apoptosis', 'melanoma', 'incidence', 'melanoma', 'continues', 'rise', 'rate', 'faster', 'cancer', 'malignant', 'melanoma', 'poor', 'prognosis', 'year', 'survival', 'rate', 'recently', 'approved', 'therapeutic', 'vemurafenib', 'extends', 'median', 'patient', 'survival', 'months', 'major', 'advance', 'raises', 'expectations', 'greater', 'rates', 'survival', 'attainable', 'combination', 'therapies']
def lemmatize_tokens(tokens):
"Gets tokens and retrieves lemmatised tokens."
# Lemmatisation using nltk lemmatiser
lmtzr = WordNetLemmatizer()
lemma = [lmtzr.lemmatize(t) for t in tokens]
return lemma
conrad2013_lemma = lemmatize_tokens(conrad2013_tokens)
huang2015_lemma = lemmatize_tokens(huang2015_tokens)
print conrad2013_lemma
['inability', 'targeted', 'braf', u'inhibitor', 'produce', 'long', 'lasting', 'improvement', 'clinical', 'outcome', 'melanoma', u'highlight', 'need', 'identify', 'additional', u'approach', 'inhibit', 'melanoma', 'growth', 'recent', u'study', 'shown', 'activation', 'wnt', 'catenin', 'pathway', u'decrease', 'tumor', 'growth', 'cooperates', 'erk', 'mapk', 'pathway', u'inhibitor', 'promote', 'apoptosis', 'melanoma', 'therefore', 'identification', 'wnt', 'catenin', u'regulator', 'advance', 'development', 'new', u'approach', 'treat', 'disease', 'order', 'goal', 'performed', 'large', 'scale', 'small', 'interfering', 'rna', 'sirna', 'screen', u'regulator', 'catenin', 'activated', 'reporter', 'activity', 'human', 'ht1080', 'fibrosarcoma', u'cell', 'integrating', 'large', 'scale', 'sirna', 'screen', 'data', 'phosphoproteomic', 'data', 'bioinformatics', 'enrichment', 'identified', 'protein', 'fam129b', 'potential', 'regulator', 'wnt', 'catenin', 'signaling', 'functionally', 'demonstrated', 'sirna', 'mediated', 'knockdown', 'fam129b', 'a375', 'a2058', 'melanoma', 'cell', u'line', 'inhibits', 'wnt3a', 'mediated', 'activation', 'catenin', 'responsive', 'luciferase', 'reporter', 'inhibits', 'expression', 'endogenous', 'wnt', 'catenin', 'target', 'gene', 'axin2', 'demonstrate', 'fam129b', 'knockdown', 'inhibits', 'apoptosis', 'melanoma', u'cell', 'treated', 'wnt3a', u'experiment', 'support', 'role', 'fam129b', 'linking', 'wnt', 'catenin', 'signaling', 'apoptosis', 'melanoma', 'incidence', 'melanoma', 'continues', 'rise', 'rate', 'faster', 'cancer', 'malignant', 'melanoma', 'poor', 'prognosis', 'year', 'survival', 'rate', 'recently', 'approved', 'therapeutic', 'vemurafenib', 'extends', 'median', 'patient', 'survival', u'month', 'major', 'advance', u'raise', u'expectation', 'greater', u'rate', 'survival', 'attainable', 'combination', u'therapy']
def bag_of_words(lemma):
"Takes in lemmatised words and returns a bow."
# Create bag of words from dictionnary
dictionary = Dictionary(lemma)
dictionary.save('../dicts/lda-text.dict')
bow = [dictionary.doc2bow(l) for l in lemma] # Calculates inverse document counts for all terms
return (bow, dictionary)
docs = [conrad2013_lemma, huang2015_lemma] # testing corpus
bow, dictionary = bag_of_words(docs)
print "Bag of words representation of the first document (tuples are composed by token_id and multiplicity):\n", bow[0]
print
for i in range(5):
print "In the document, topic_id %d (word %s) appears %d time[s]" %(bow[0][i][0], docs[0][bow[0][i][0]], bow[0][i][1])
print "..."
Bag of words representation of the first document (tuples are composed by token_id and multiplicity): [(0, 1), (1, 1), (2, 1), (3, 2), (4, 1), (5, 1), (6, 2), (7, 3), (8, 2), (9, 1), (10, 1), (11, 1), (12, 1), (13, 1), (14, 1), (15, 7), (16, 3), (17, 1), (18, 1), (19, 1), (20, 1), (21, 2), (22, 1), (23, 1), (24, 1), (25, 1), (26, 1), (27, 1), (28, 1), (29, 1), (30, 1), (31, 1), (32, 1), (33, 1), (34, 4), (35, 1), (36, 1), (37, 1), (38, 1), (39, 1), (40, 1), (41, 2), (42, 1), (43, 1), (44, 1), (45, 1), (46, 1), (47, 1), (48, 1), (49, 1), (50, 1), (51, 1), (52, 2), (53, 3), (54, 1), (55, 1), (56, 2), (57, 2), (58, 1), (59, 1), (60, 1), (61, 1), (62, 1), (63, 1), (64, 1), (65, 1), (66, 1), (67, 2), (68, 8), (69, 1), (70, 1), (71, 1), (72, 1), (73, 1), (74, 2), (75, 1), (76, 1), (77, 1), (78, 1), (79, 1), (80, 1), (81, 1), (82, 1), (83, 1), (84, 1), (85, 3), (86, 1), (87, 1), (88, 3), (89, 2), (90, 1), (91, 1), (92, 1), (93, 1), (94, 2), (95, 2), (96, 1), (97, 2), (98, 3), (99, 1), (100, 1), (101, 1), (102, 3), (103, 1), (104, 1), (105, 1), (106, 1), (107, 1), (108, 1), (109, 1), (110, 1), (111, 1), (112, 5), (113, 2), (114, 1)] In the document, topic_id 0 (word inability) appears 1 time[s] In the document, topic_id 1 (word targeted) appears 1 time[s] In the document, topic_id 2 (word braf) appears 1 time[s] In the document, topic_id 3 (word inhibitor) appears 2 time[s] In the document, topic_id 4 (word produce) appears 1 time[s] ...
def lda_model(dictionary, bow):
ldamodel = models.ldamodel.LdaModel(bow, num_topics=10, id2word=dictionary, passes=5)
return ldamodel
lda = lda_model(dictionary, bow)
for index, score in sorted(lda[bow[1]], key=lambda tup: -1*tup[1]):
print "Score: %f\nTopic: %s" %(score, lda.print_topic(index, 10))
WARNING:gensim.models.ldamodel:too few updates, training might not converge; consider increasing the number of passes or iterations to improve accuracy
Score: 0.990722 Topic: 0.080*gnt1ip + 0.027*golgi + 0.027*mgat1 + 0.018*activity + 0.018*reduced + 0.018*inhibitory + 0.018*interaction + 0.018*fret + 0.018*expressed + 0.018*medial
# Try the model on another article abstract:
burns2015 = """Duplication of the yeast centrosome (called the spindle pole body, SPB) is thought to occur through a series of discrete steps that culminate in insertion of the new SPB into the nuclear envelope (NE).
To better understand this process, we developed a novel two-color structured illumination microscopy with single-particle averaging (SPA-SIM) approach to study the localization of all 18 SPB components during duplication using endogenously expressed fluorescent protein derivatives.
The increased resolution and quantitative intensity information obtained using this method allowed us to demonstrate that SPB duplication begins by formation of an asymmetric Sfi1 filament at mitotic exit followed by Mps1-dependent assembly of a Spc29- and Spc42-dependent complex at its tip.
Our observation that proteins involved in membrane insertion, such as Mps2, Bbp1, and Ndc1, also accumulate at the new SPB early in duplication suggests that SPB assembly and NE insertion are coupled events during SPB formation in wild-type cells."""
burns2015_tokens = lemmatize_tokens(get_tokens(parse_text(burns2015)))
bow_vector = dictionary.doc2bow(burns2015_tokens)
for index, score in sorted(lda[bow_vector], key=lambda tup: -1*tup[1]):
print "Score: %f\n Topic: %s" %(score, lda.print_topic(index, 5))
Score: 0.610076 Topic: 0.044*melanoma + 0.039*catenin + 0.028*wnt + 0.023*fam129b + 0.017*cell Score: 0.328385 Topic: 0.080*gnt1ip + 0.027*golgi + 0.027*mgat1 + 0.018*activity + 0.018*reduced
def prepare_text_for_lda(text):
# Clean
doc = [text.replace(unicode(i), spe_char.get(i)) for i in text if i in spe_char.keys()] or [text]
# Tokenise
texts = doc[0].lower().replace('\n', ' ').replace('/', ' ').replace('-', ' ').split(' ')
# Remove punctuation and stop words
tokens = [filter(lambda x:x not in string.punctuation, i)
for i in texts if i != '' and i not in STOPWORDS]
tokens_cleaned = [i for i in tokens if len(i) > 2 and not i.isdigit()]
# Lemmatise
lmtzr = WordNetLemmatizer()
lemmatised_text = [lmtzr.lemmatize(t) for t in tokens_cleaned]
return lemmatised_text
def parse_from_file(text_file):
"Retrieves the content of a text file"
with codecs.open(text_file, mode='r', encoding='utf-8') as f:
reader = f.read()
return reader
from os import listdir
from os.path import isfile, join
# Gather the articles
filepath = "../articles/Cell biology/"
articles = [f for f in listdir(filepath) if isfile(join(filepath, f))] or []
print "There are %d articles in this folder.\n" % len(articles)
# Create a corpus
corpus = [prepare_text_for_lda(parse_from_file(filepath + a)) for a in articles[:45]]
articles_bow, articles_dict = bag_of_words(corpus)
articles_lda = lda_model(articles_dict, articles_bow)
for i in range(5):
for index, score in sorted(articles_lda[articles_bow[i]], key=lambda tup: -1*tup[1]):
print "Article %s\n Score: %f - Topic: %s\n" %(articles[i], score, articles_lda.print_topic(index, 10))
WARNING:gensim.models.ldamodel:too few updates, training might not converge; consider increasing the number of passes or iterations to improve accuracy
There are 67 articles in this folder. Article elife00772.txt Score: 0.999293 - Topic: 0.054*cell + 0.009*protein + 0.008*figure + 0.008*pom1 + 0.007*gfp + 0.005*membrane + 0.005*pom1p + 0.005*strain + 0.005*time + 0.005*intensity Article elife00654.txt Score: 0.999173 - Topic: 0.020*protein + 0.019*cell + 0.012*membrane + 0.007*cavin + 0.006*figure + 0.006*ap2 + 0.006*cargo + 0.006*ift + 0.005*function + 0.005*cilium Article elife02137.txt Score: 0.863058 - Topic: 0.022*cell + 0.011*protein + 0.010*cenp + 0.010*redox + 0.010*aging + 0.008*figure + 0.008*oxidation + 0.008*nadph + 0.008*yeast + 0.007*level Article elife02137.txt Score: 0.082134 - Topic: 0.020*protein + 0.019*cell + 0.012*membrane + 0.007*cavin + 0.006*figure + 0.006*ap2 + 0.006*cargo + 0.006*ift + 0.005*function + 0.005*cilium Article elife02137.txt Score: 0.053982 - Topic: 0.054*cell + 0.009*protein + 0.008*figure + 0.008*pom1 + 0.007*gfp + 0.005*membrane + 0.005*pom1p + 0.005*strain + 0.005*time + 0.005*intensity Article elife03083.txt Score: 0.998843 - Topic: 0.054*cell + 0.009*protein + 0.008*figure + 0.008*pom1 + 0.007*gfp + 0.005*membrane + 0.005*pom1p + 0.005*strain + 0.005*time + 0.005*intensity Article elife06156.txt Score: 0.998976 - Topic: 0.029*cell + 0.029*cln3 + 0.014*length + 0.013*whi5 + 0.012*start + 0.011*1—figure + 0.011*supplement + 0.011*decision + 0.009*signal + 0.009*http
print "Cell biol article:"
cb_article = articles[50]
print cb_article
cb_tokens = prepare_text_for_lda(parse_from_file(filepath + cb_article))
cb_bow = articles_dict.doc2bow(cb_tokens)
for index, score in sorted(articles_lda[cb_bow], key=lambda tup: -1*tup[1]):
print "\nScore: %f\nTopic: %s\n" %(score, articles_lda.print_topic(index, 10))
print "-" * 70
print "Neuro article:"
filepath_ns = "../articles/Neuroscience/"
articles_ns = [f for f in listdir(filepath_ns) if isfile(join(filepath_ns, f))] or []
ns_article = articles_ns[0]
print ns_article
ns_tokens = prepare_text_for_lda(parse_from_file(filepath_ns + ns_article))
ns_bow = articles_dict.doc2bow(ns_tokens)
for index, score in sorted(articles_lda[ns_bow], key=lambda tup: -1*tup[1]):
print "\nScore: %f\nTopic: %s" %(score, articles_lda.print_topic(index, 10))
Cell biol article: elife03307.txt Score: 0.999176 Topic: 0.054*cell + 0.009*protein + 0.008*figure + 0.008*pom1 + 0.007*gfp + 0.005*membrane + 0.005*pom1p + 0.005*strain + 0.005*time + 0.005*intensity ---------------------------------------------------------------------- Neuro article: elife05438.txt Score: 0.364755 Topic: 0.020*protein + 0.019*cell + 0.012*membrane + 0.007*cavin + 0.006*figure + 0.006*ap2 + 0.006*cargo + 0.006*ift + 0.005*function + 0.005*cilium Score: 0.336101 Topic: 0.054*cell + 0.009*protein + 0.008*figure + 0.008*pom1 + 0.007*gfp + 0.005*membrane + 0.005*pom1p + 0.005*strain + 0.005*time + 0.005*intensity Score: 0.111372 Topic: 0.028*micos + 0.016*complex + 0.016*protein + 0.015*cell + 0.015*qil1 + 0.013*mitochondrial + 0.012*figure + 0.011*membrane + 0.010*cristae + 0.010*subunit Score: 0.055396 Topic: 0.016*usp13 + 0.014*protein + 0.014*chac1 + 0.012*glutathione + 0.011*expression + 0.011*usp5 + 0.008*tagged + 0.007*activity + 0.007*figure + 0.006*mouse Score: 0.046261 Topic: 0.029*cell + 0.029*cln3 + 0.014*length + 0.013*whi5 + 0.012*start + 0.011*1—figure + 0.011*supplement + 0.011*decision + 0.009*signal + 0.009*http Score: 0.036583 Topic: 0.046*snap + 0.031*cell + 0.027*alpha + 0.022*rnai + 0.017*soce + 0.013*orai1 + 0.012*stim1 + 0.009*calcium + 0.009*figure + 0.009*1—figure Score: 0.030731 Topic: 0.012*cell + 0.009*alpha + 0.008*bpac + 0.008*tnf + 0.007*figure + 0.007*protein + 0.007*control + 0.006*cftr + 0.005*expression + 0.005*golgi Score: 0.018469 Topic: 0.022*cell + 0.011*protein + 0.010*cenp + 0.010*redox + 0.010*aging + 0.008*figure + 0.008*oxidation + 0.008*nadph + 0.008*yeast + 0.007*level
1- Who I am
2- Goal of this project
3- My plan
4- First iteration
5- Improvements
6- Second iteration
7- Discussion
Discussion¶
State of the project:
LDA seems like a good tool to compare articles on their content
Next step: train lda model on different types of articles
Idea: use the number of topics to evalute the proximity with a subject
Tools used:
- Python 2.7 and iPython notebook
- Nltk library for natural text processing (nlp)
- Gensim library for nlp and topic modeling
- Scikit-learn library for Machine Learning
That's all folks!¶
####Questions and suggestions welcome =)
####@EleonoreMayola
github.com/Eleonore9/get-articles-meaning