Dealing with Bias and Variance¶
import addutils.toc ; addutils.toc.js(ipy_notebook=True)
import scipy.io
import numpy as np
import pandas as pd
from time import time
from addutils import css_notebook
from sklearn import metrics
import sys
import os
from IPython.core.display import Image
css_notebook()
1 Bias-Variance Tradeoff¶
1.1 (In)Formal introduction to Bias and Variance¶
Roughly speaking Machine Learning algorithms try to characterize a tradeoff between generalization (how well the model represents the general concept or function to learn) and apporoximation (how well the model fits the data). A small error denotes a good approximation of the function out of sample. A more complex model has a better chance of approximating $f$, in contrast a less complex model has a better chance of generalizing (picking the right approximation) out of sample.
A more formal definition can be given after Hastie, et al. 2009. Let's explain Bias-Variance using linear regression problems, and specifically we'll use a squared-error measure as loss function. Assume further that the output $Y = f(X) + \epsilon$ where $E(\epsilon) = 0$ and $Var(\epsilon) = \sigma_\epsilon^2$
We estimate a model $\widehat{f}(X)$ of a function $f(X)$, and the expected squared error of a given point $x_0$ is:
$$Err(x_0) = E[(Y - \widehat{f}(x_0))^2]$$It is possible to decompose this error in its Bias and Variance components:
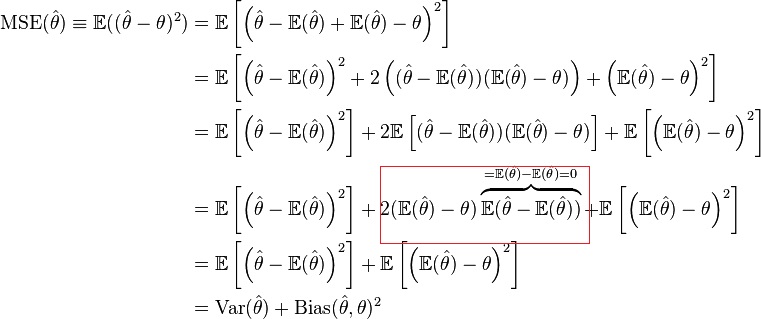
Where $Irreducible Error$ is the variance in the true relationship that cannot be captured by any model no matter how well we estimate $f(X)$. $Bias^2$ is the difference between the average of our estimate from the true mean. $Variance$ is the expected squared deviation of $\widehat{f}(x_0)$ around its mean. Usually the more complex is the model the lower the Bias but the higher the variance. We can give a pictorial illustration of the relationship with the image below. (The image is taken from this blog)
Image('images/bias_variance.png')

Suppose the center (red) of each diagram is the real function to learn. As we move away from the center the prediction gets worse and worse. Each dot in the picture is a realization of the model given a subsample of the dataset; each circle is a different model (whose particular realizations are the dots) and model's characteristics are described by the two corresponding labels (horizontal and vertical).
Having a model (top left) with both Low Bias and Low Variance is practically impossible since, as we will see next, if we decrese one the other increase (in this case both generalization and approximation will be good and we could learn almost the real function); the hard part is hence finding the best possible balance between the two. In case of Low Bias and High Variance (top right), the model is capable of representing the correct function but the model may be too complex, and thus more supsceptible in variation on the dataset. For this reason each dot (function approximation) draw from this model may vary considerably from estimate to estimate, depending on the particular subsample used for training. For High Bias and Low Variance (bottom left) the model is constantly learning a substantially different function respect to the true one, but each run is independent from the particular subsample of data (it generalize well but it approximate poorly). If we have a model that has both High Varianche and High Bias (bottom right) the model si constantly learning a wrong model with high variability between subsamples and thus the probability of picking a good model is very low.
One can think of Bias and Variance as urn that contains a bunch of candidate approximations of an unknown function $f$. If we have few candidates in the urn (think about degenerating and have only one function in it), but each candidate is far away from the true value of $f$; in this case the Bias is high and the Variance is low, no matter of the dataset we will always return the same function. If we have a bigger urn containing a lot of candidates the probability that it contains the true function is higher, but it is difficult to navigate through the candidates to find the right one. In this case the Bias is lower but the Variance is higher, we have so many varieties that depending on which sample we get the function differs.
Next we will show what we mean by complexity of the model with some practical examples in order to further understand the Bias-Variance tradeoff.
from utilities import biasvariance
from bokeh.models.ranges import Range1d
In this figure, we use polynomials with different degrees d to fit the same data.
For d=1, the model suffers from high bias: the model complexity is not enough to represent the data.
At the other extreme, for d=6 the model suffers from high variance: the mode overfit the data because it has too many free parameters. In this case a typical behaviour is that if any of the input points are varied slightly, it could result in a very different approximation. This kind of model will fit perfectly the training data while failing to do so on validation data.
In the middle picture, for d=2, we have found a good model.
%matplotlib inline
import matplotlib.pyplot as plt
biasvariance.plot_bias_variance(8, random_seed=42, err=10)
1.2 Bias-Variance Tradeoff explained with a regression example¶
In this section we will explore a simple linear regression problem.
This can be accomplished within scikit-learn with the sklearn.linear_model module.
We consider the situation where we have only 2 data points:
from sklearn import linear_model
X = np.array([0.5, 1.0]).reshape(2,1)
y = np.array([0.5, 1.0])
X_test = np.array([0.0, 2.0]).reshape(2,1)
regr = linear_model.LinearRegression()
regr.fit(X, y)
fig = bk.figure(plot_width=400, plot_height=300, title=None)
fig.circle(X[:,0], y, size=5)
fig.line(X_test[:,0], regr.predict(X_test), color='green')
bk.show(fig)
In real life situation, we have noise (e.g. measurement noise) in our data:
import seaborn as sns
import addutils.palette as pal
import random
cat_colors = list(map(pal.to_hex, sns.color_palette('bright', 6)))
random.shuffle(cat_colors)
np.random.seed(0)
fig = bk.figure(plot_width=400, plot_height=300, title=None)
for i in range(6):
noise = np.random.normal(loc=0, scale=.1, size=X.shape)
noisy_X = X + noise
regr.fit(noisy_X, y)
fig.circle(noisy_X[:,0], y, size=8, fill_color=cat_colors[i], line_color='black')
fig.line(X_test[:,0], regr.predict(X_test), color=cat_colors[i])
bk.show(fig)
As we can see, our linear model captures and amplifies the noise in the data. It displays a lot of variance. This means that if we subsample the data and we fit a different model to each subsample (each pair of points) we obtain a model that differ substantially from the true model. There are several techniques to reduce variance. Regularization is one of those, it is a way to change the tradeoff between Bias and Variance by putting more weights on either one. We will briefly use it in this example while we leave a more detailed explanation for the next section.
We can use another linear estimator that uses regularization: the Ridge estimator. This estimator regularizes the coefficients by shrinking them to zero, under the assumption that very high correlations are often spurious. High alphas give high regularization (shrinkage):
regr = linear_model.Ridge(alpha=0.08)
fig = bk.figure(plot_width=400, plot_height=300, title=None)
for i in range(6):
noise = np.random.normal(loc=0, scale=.1, size=X.shape)
noisy_X = X + noise
regr.fit(noisy_X, y)
fig.circle(noisy_X[:,0], y, size=8, color=cat_colors[i], line_color='black')
fig.line(X_test[:,0], regr.predict(X_test), color=cat_colors[i])
bk.show(fig)
As we can see, the estimator displays much less variance. However it systematically under-estimates the coefficient. It displays a biased behavior. As explained earlier we changed the tradeoff between Bias and Variance with regularization. Changing one quantity (for example decreasing Variance) inevitably increases the other (Bias).
With the next examples we will try to answer the following question: If our estimator is underperforming, how should we move forward?
- Do I need a Simple or more Complicated Model ?
- Do I need More Training Samples ?
- Do I need more features for each observed data point ?
The answer is often counter-intuitive. In particular, Sometimes using a more complicated model will give worse results. Also, Sometimes adding training data will not improve your results. The ability to determine what steps will improve your model is what separates the successful machine learning practitioners from the unsuccessful.
2 Regularization: what it is and why it is necessary¶
The core idea behind regularization is that we are going to prefer models that are simpler, even if they lead to more errors on the training set. We start by defining a 9th order polynomial function. This represents our 'ground truth'. You can imagine it like a signal measured at diffrent times:
f = lambda t: 1.2*t**2 + 0.1*t**3 - 0.6*t**5 - 0.8*t**9
gt_coeff = [0, 0, 1.2, 0.1, 0., -0.6, 0., 0., 0., -0.8]
Unfortunately in real life situation every signal is affected by a measurement error; in this example we simulate it with the variable noise_level. Our ground truth is a 9th order polynomial.
At first it would seem an obvius choice to use a 9th order polynomial to fit the signal. If you play a little with the following code you will discover that not using a regularization technique (LinearRegression doesn't allow any regularization), most of the time could be a bad choice, and a simpler (lower-order) model it is much better to avoid overfitting.
Try to change the following variables:
orders: orders of the polynomials to fitn_samples: when the number of the samples is small it's difficult to fit a high-order model and you have overfittingnoise_level: whit very low noise it's easier to fit higher-order polynomials
Keep in mind that we didn't fix the random generator seed, so every time you run the cell you'll have a different noise distribution on the samples.
As you can see we use a linear algorithm to fit a nonlinear function, this is possible because we use the linear algorithm to fit the nonlinear coefficients that we define in the regressors.
tmin, tmax = -1.1, 1.0
n_samples = 25
noise_level = 0.2
orders = [4, 9]
np.random.seed(1)
t = tmin + np.random.rand(n_samples) * (tmax-tmin) # Sample points
y_noisy = f(t) + noise_level*np.random.normal(size=n_samples) # Noisy measure
fig = bk.figure(plot_width=700, plot_height=400,
title='4th and a 9th order polynomial with Ground truth')
fig.circle(t, y_noisy, size=8, fill_alpha=0.4)
p = np.linspace(tmin, tmax, 200) # Array to calc. the prediction
colors = ['royalblue', 'green']
for order in orders:
X = np.array([t**i for i in range(order+1)]).T # Regressor
Xp = np.array([p**i for i in range(order+1)]).T # Regressor for prediction
poly_linreg = linear_model.LinearRegression().fit(X, y_noisy)
fig.line(p, poly_linreg.predict(Xp), legend='linreg order: %02i' % order,
color=colors.pop(), line_width=3.0)
fig.line(p, f(p), legend="truth", color='red', line_width=4.0)
fig.legend.label_text_font_size = '14pt'
fig.legend.location = 'top_left'
np.set_printoptions(precision=2)
print('Ground Truth coeff.: ', ' '.join(['%+5.1f' %n for n in gt_coeff]))
print('LinReg coefficients: ', ' '.join(['%+5.1f' %n for n in poly_linreg.coef_]))
bk.show(fig)
Ground Truth coeff.: +0.0 +0.0 +1.2 +0.1 +0.0 -0.6 +0.0 +0.0 +0.0 -0.8 LinReg coefficients: +0.0 -0.9 +4.3 +11.6 -18.2 -42.8 +35.3 +61.7 -21.4 -31.9
Now we compare the previous LinearRegression algorithm with the Lasso algorithm.
Lasso (least absolute shrinkage and selection operator) is an alternative regularized version of least squares: it is a shrinkage estimator: unlike ridge regression, as the penalty term increases, lasso sets more coefficients to zero, this means that the lasso estimator produces smaller models.
order = 9
np.random.seed(1)
X = np.array([t**i for i in range(order+1)]).T
Xp = np.array([p**i for i in range(order+1)]).T
poly_linreg = linear_model.LinearRegression().fit(X, y_noisy)
poly_lasso = linear_model.Lasso(alpha = 0.005).fit(X, y_noisy)
fig = bk.figure(plot_width=700, plot_height=400)
fig.circle(t, y_noisy, size=8, alpha=0.4)
fig.line(p, poly_linreg.predict(Xp), legend='linreg order: %02i' % order,
color='royalblue', line_width=3.0)
fig.line(p, poly_lasso.predict(Xp), legend='lasso order: %02i' % order,
line_width=3.0, color='green')
fig.line(p, f(p), legend="truth", color='red', line_width=4.0)
fig.legend.label_text_font_size = '15pt'
fig.legend.location = 'top_left'
fig.title.text = '9th order LinReg and Lasso with Ground truth'
np.set_printoptions(precision=2)
print('Ground Truth coeff.: ', ' '.join(['%+5.1f' %n for n in gt_coeff]))
print('LinReg coefficients: ', ' '.join(['%+5.1f' %n for n in poly_lasso.coef_]))
bk.show(fig)
Ground Truth coeff.: +0.0 +0.0 +1.2 +0.1 +0.0 -0.6 +0.0 +0.0 +0.0 -0.8 LinReg coefficients: +0.0 -0.0 +1.0 -0.0 +0.0 -0.0 +0.0 -0.0 +0.0 -1.3
Try by yourself: play a little with the code of the two previous cells by changing n_samples and noise_level to verify that:
- When
n_samplesis very high (>1000) it's very unlikely to have overfitting. This means that with more data it's possible to fit more complex models without overfitting. - When
n_samplesis very low (<40) it's almost impossible to fit with Linear Regression without overfitting. In this case regularization is always required - When
n_samplesis very high (>1000) andnoise_levelis very low, the Lasso algorithm keeps just the coefficients actually used to calculate the ground truth function. This means that Lasso can deal with high dimesional problems where most of the features can be neglected producing compact linear models.
Another way to look at the Bias-Variance tradeoff in previous examples is to consider how the model fits the data. As stated at the beginning of this section when model complexity increases Bias decrease while Variance increses. The optimal model performance is achieved when the level of complexity is such that an increase in Bias is equivalent in reduction of Variance. If the model complexity exceed this threshold we are overfitting (the model is to adapted to the data and does not generalize well) if the model is behind this threshold is said to be underfitting (the model does not fit well the data and it has a poor error for out of sample data).
3 Separate Training and Validation Sets¶
Fitting a model and testing it on the same data is a methodological mistake: a model could have a perfect score but would fail to predict anything useful on yet-unseen data. This situation is called overfitting. To avoid it, it is common practice to hold out part of the available data as a Validation Set X_valid, y_valid.
from sklearn import datasets, preprocessing, metrics
X, y = datasets.samples_generator.make_regression(n_samples=70,
n_features=1, n_informative=1,
random_state=0, noise=5)
scaler = preprocessing.MinMaxScaler()
X_sc = scaler.fit_transform(X)
lin = linear_model.LinearRegression(fit_intercept=True)
lin.fit(X_sc, y)
print(lin)
print("Model coefficient: %.5f, and intercept: %.5f" % (lin.coef_, lin.intercept_))
err = metrics.mean_squared_error(lin.predict(X_sc), y)
print("Mean Squared Error: %.2f" % err)
LinearRegression(copy_X=True, fit_intercept=True, n_jobs=1, normalize=False) Model coefficient: 275.99550, and intercept: -145.41186 Mean Squared Error: 23.04
In scikit-learn a random split into Training and Validation sets can be quickly computed with the train_test_split helper function:
from sklearn import model_selection
X_tr, X_valid, y_tr, y_valid = model_selection.train_test_split(X_sc, y,
test_size=0.30,
random_state=0)
lin.fit(X_tr, y_tr)
MSE = metrics.mean_squared_error(lin.predict(X_valid), y_valid)
print("Mean Squared Error: %.2f" % MSE)
print("Model coefficient: %.5f, and intercept: %.5f" % (lin.coef_, lin.intercept_))
X_predicted = np.linspace(0, 1, 2)[:, np.newaxis]
y_predicted = lin.predict(X_predicted)
fig = bk.figure(title='Training and Validation Sets',
plot_width=700, plot_height=400)
fig.circle(X_tr.squeeze(), y_tr,
size=5, line_alpha=0.85, fill_color='green', line_color='darkgreen',
legend='Training Set')
fig.circle(X_valid.squeeze(), y_valid,
size=5, line_alpha=0.85, fill_color='orangered', legend='Validation Set')
fig.line(X_predicted.ravel(), y_predicted, line_color='blue', legend='Linear Regression')
fig.legend.location = 'bottom_right'
bk.show(fig)
Mean Squared Error: 23.34 Model coefficient: 273.00288, and intercept: -143.70277
3.1 Example: Do a Regression Analysis on MATLAB® data¶
The data are generated with the following MATLAB code:
% Generate Regression Test Data 02 ----------------------------------------
samples = 100; features = 5;
X = rand(samples, features)
for i = 1:samples
X(i,:) = (X(i,:)*i)+i+0.1;
end
% Calculate y as a linear combination of features with coeff. 1.5, 2.5, ...
% and add some noise
noise = 0.1;
lin_comb = (1:features) + 0.5
y = (X+rand(samples, features)*noise)*lin_comb'
% Define feature names as 'F001', ... , 'Fnnn' up to 9999 features
feat_names = sprintf('F%04i',1);
for i = 2:features
feat_names = strvcat(feat_names, sprintf('F%04i',i));
end
save ('matlab_test_data_02', 'X','y', 'feat_names')
from sklearn import preprocessing, linear_model
mat = scipy.io.loadmat('example_data/matlab_test_data_02.mat')
col = [s.strip() for s in list(mat['feat_names'])]
Xt,Xv,yt,yv = model_selection.train_test_split(mat['X'], mat['y'],test_size=0.30,
random_state=0)
Xt = pd.DataFrame(Xt, columns=col)
Xv = pd.DataFrame(Xv, columns=col)
yt = pd.DataFrame(yt, columns=['measured'])
yv = pd.DataFrame(yv, columns=['measured'])
Xt.head(3)
| F0001 | F0002 | F0003 | F0004 | F0005 | F0006 | |
|---|---|---|---|---|---|---|
| 0 | 131.391611 | 178.588071 | 191.836028 | 206.928031 | 188.601869 | 195.879822 |
| 1 | 137.771630 | 91.462643 | 76.438656 | 104.585583 | 123.265090 | 73.587835 |
| 2 | 789.737369 | 671.934539 | 835.830887 | 515.921167 | 901.167154 | 813.659391 |
# Scaler is fitted only on training data
Xts = Xt.copy()
Xvs = Xv.copy()
scaler = preprocessing.StandardScaler(copy=False, with_mean=True, with_std=True).fit(Xt)
scaler.transform(Xts)
scaler.transform(Xv)
array([[ 2.46, 0.88, 1.49, 2.39, 2.1 , 2.5 ],
[ 0.5 , 0.64, 0.64, 0.54, 0.43, 0.96],
[-0.75, -0.93, -0.81, -0.61, -0.44, -0.83],
...,
[ 1.24, 2.17, 0.7 , 2.22, 1.78, 2.63],
[ 0.4 , -0.15, -0.38, 0.05, -0.17, 0.4 ],
[-1.53, -1.55, -1.55, -1.55, -1.58, -1.55]])
Xts.head(3)
| F0001 | F0002 | F0003 | F0004 | F0005 | F0006 | |
|---|---|---|---|---|---|---|
| 0 | -1.294851 | -1.210162 | -1.171325 | -1.148142 | -1.198971 | -1.160278 |
| 1 | -1.281377 | -1.399557 | -1.416333 | -1.371187 | -1.344097 | -1.424161 |
| 2 | 0.095500 | -0.137716 | 0.195982 | -0.474723 | 0.383782 | 0.172776 |
model = linear_model.LinearRegression(fit_intercept=True, normalize=True)
model.fit(Xts, yt)
# NOTICE: coefficients are much different from 1.5, 2.5, ... when fitting on scaled data
print(model.coef_)
print(model.intercept_)
[[ 708.82 1153.81 1647.24 2066.73 2474.24 3011.71]] [17692.81]
# Plot the data and the model prediction
yp = model.predict(Xvs)
fig = bk.figure(title='Measured Values VS Predicted Values',
x_axis_label='y val', y_axis_label='y pred',
plot_width=600, plot_height=400)
fig.circle(yv['measured'], yp[:,0], size=8,
fill_color='blue', fill_alpha=0.5, line_color='black', alpha=0.2)
bk.show(fig)
3.2 Example: Training and a Validation Sets on a Classification Problem¶
The K-neighbors is an instance-based classifier that predicts the label of an unknown point based on the labels of the K nearest points in the parameter space:
from sklearn import neighbors
digits = datasets.load_digits()
X, y = digits.data, digits.target
clf = neighbors.KNeighborsClassifier(n_neighbors=1)
clf.fit(X, y)
y_pred = clf.predict(X)
print(metrics.confusion_matrix(y_pred, y))
print('\nF1 Score: ', metrics.f1_score(y_pred, y, average='weighted'))
[[178 0 0 0 0 0 0 0 0 0] [ 0 182 0 0 0 0 0 0 0 0] [ 0 0 177 0 0 0 0 0 0 0] [ 0 0 0 183 0 0 0 0 0 0] [ 0 0 0 0 181 0 0 0 0 0] [ 0 0 0 0 0 182 0 0 0 0] [ 0 0 0 0 0 0 181 0 0 0] [ 0 0 0 0 0 0 0 179 0 0] [ 0 0 0 0 0 0 0 0 174 0] [ 0 0 0 0 0 0 0 0 0 180]] F1 Score: 1.0
Apparently, we've found a perfect classifier! But this is misleading for the reasons we saw before: the classifier essentially "memorizes" all the samples it has already seen. To really test how well this algorithm does, we need to try some samples it hasn't yet seen.
Here we split the original data in Training and Validation sets and run again the previous algorithm. In this case we see that we still have a good classifier but the precision is reduced:
X_tr, X_valid, y_tr, y_valid = model_selection.train_test_split(X,
y,
test_size=0.40,
random_state=0)
clf = neighbors.KNeighborsClassifier(n_neighbors=1).fit(X_tr, y_tr)
y_pred = clf.predict(X_valid)
print(metrics.confusion_matrix(y_valid, y_pred))
print('\nF1 Score: ', metrics.f1_score(y_valid, y_pred, average='weighted'))
[[60 0 0 0 0 0 0 0 0 0] [ 0 73 0 0 0 0 0 0 0 0] [ 0 0 71 0 0 0 0 0 0 0] [ 0 0 0 70 0 0 0 0 0 0] [ 0 0 0 0 63 0 0 0 0 0] [ 0 0 0 0 0 87 1 0 0 1] [ 0 0 0 0 0 0 76 0 0 0] [ 0 0 0 0 0 0 0 65 0 0] [ 0 2 0 1 0 0 0 0 74 1] [ 0 0 0 2 0 1 0 0 0 71]] F1 Score: 0.9874410807522929
4 Do I need a Simple or a Complex Model?¶
In order to show the relation between the size of the training dataset and the complexity of the model we generate some example data in the same way we did in the very first example but with more than ten times the number of samples. The we fit a polynomial model and we plot the error w.r.t. the degree of polynomial.
Here Training Data are plotted in Blue while Validation Data are Red:
from sklearn import model_selection
N = 100
test_size = 0.40
error = 1.0
np.random.seed(1)
x = np.random.random(N)
y = biasvariance.test_func(x, error)
x_tr, x_valid, y_tr, y_valid = model_selection.train_test_split(x, y, test_size=test_size)
fig = bk.figure(plot_width=700, plot_height=400, title=None)
fig.circle(x_tr, y_tr, color='blue', size=6, alpha=0.5)
fig.circle(x_valid, y_valid, color='red', size=6, alpha=0.5)
bk.show(fig)
The model parameters (in our case, the coefficients of the polynomials) are learned using the training set.
The meta-parameters (in our case, the degree of the polynomial) are adjusted so that this validation error is minimized.
Finally, the labels are predicted for the test set.
The validation error of our polynomial classifier can be visualized by plotting the error as a function of the polynomial degree:
import warnings
warnings.filterwarnings('ignore', message='Polyfit*')
degrees = np.arange(41)
train_err = np.zeros(len(degrees))
validation_err = np.zeros(len(degrees))
for i, d in enumerate(degrees):
p = np.polyfit(x_tr, y_tr, d)
train_err[i] = biasvariance.compute_error(x_tr, y_tr, p)
validation_err[i] = biasvariance.compute_error(x_valid, y_valid, p)
fig = bk.figure(plot_width=700, plot_height=400, title=None)
fig.line(degrees, validation_err,
line_width=2, legend='cross-validation error', color='royalblue')
fig.line(degrees, train_err, line_width=2, legend='training error', color='green')
fig.line([0, 20], [error, error], line_dash='dashed', legend='intrinsic error', color='black')
fig.xaxis.axis_label = 'degree of fit'
fig.yaxis.axis_label = 'rms error'
fig.grid.grid_line_color = None
bk.show(fig)
For this toy dataset, error = 1.0 is the best we can hope to obtain. Choosing d=6 in this case gets us very close to the optimal error.
Notice that in the above plot, d=6 gives the best results. But in the very first example, we found that d=6 vastly over-fits the data. What’s going on here? The difference is the number of training points used:
As a general rule of thumb, the more the training points, the more complex the model can be.
5 Do I need More Training Samples?¶
In order to exploit the relationship outlined above, between the complexity of the model and the number of examples, we use a tool called Learning Curves. Learning Curves works by plotting the training error and validation (or test) error as a function of the number of training points:
def plot_learning_curve(d, N=100, y_range=None):
test_size = 0.40
n_sizes = 50 # Number of testing point in which to split the size
n_runs = 20 # Number of times to run the test for each training set size
sizes = np.linspace(2, N, n_sizes).astype(int)
train_err = np.zeros((n_runs, n_sizes))
validation_err = np.zeros((n_runs, n_sizes))
for run in range(n_runs):
for nsize, size in enumerate(sizes):
x_tr, x_valid, y_tr, y_valid = model_selection.train_test_split(x, y,
test_size=test_size, random_state=run)
# Train on only the first `size` points
p = np.polyfit(x_tr[:size], y_tr[:size], d)
# Validation error is on the *entire* validation set
validation_err[run, nsize] = biasvariance.compute_error(x_valid, y_valid, p)
# Training error is on only the points used for training
train_err[run, nsize] = biasvariance.compute_error(x_tr[:size], y_tr[:size], p)
fig = bk.figure(plot_width=400, plot_height=300, title='d = %i' % d,
x_range=(0, N-1))
fig.title.text_font_size = '11pt'
fig.xaxis.axis_label_text_font_size = '9pt'
fig.yaxis.axis_label_text_font_size = '9pt'
fig.line(sizes, validation_err.mean(axis=0),
line_width=2, legend='mean validation error', color='royalblue')
fig.line(sizes, train_err.mean(axis=0),
line_width=2, legend='mean training error', color='green')
fig.line([0, N], [error, error],
line_dash='dashed', legend='intrinsic error', color='black')
fig.xaxis.axis_label = 'traning set size'
fig.yaxis.axis_label = 'rms error'
fig.legend.location = 'top_right'
if y_range:
fig.y_range = Range1d(y_range[0], y_range[1])
fig.grid.grid_line_color = None
bk.show(fig)
Now we look at the behavior of $E_{train}$ and $E_{test}$.
As we increase $N$, $E_{test}$ decreases until the limit of the algorithm, that is its $Bias$.
As we increase $N$, $E_{train}$ increases, because with few datapoints the task is simpler, while adding more data increase the difficulty of the algorithm to fit them.
The discrepancy of the two curves is the generalization error. It should shrink and get tighter as generalization increases.
Image('images/learning_curves.png')

In the left picture (simple model) we can see that the model "true" error (the best the model can do) is high and pretty soon (that is for lower values of $N$) we reach the point were both $E_{train}$ and $E_{test}$ are closer. In the right picture (complex model) we can see that, since it is more complex, it has a better approximation of the target function and thus can achieve (in principle) a lower $E_{test}$ error. The function is so complex that with few data points it fits them perfectly reaching a training error of zero. However if we look at the corresponding $E_{test}$ we can see that it is very high, we haven't learn anything. As the number of examples grows, $E_{test}$ starts lowering and it gets lower than the other model, but the discrepancy between the two errors remains higher than that of the previous example.
Let's see a practical example:
plot_learning_curve(d=1, N=100)
A polynomial model with d=1 is a high-biased estimator which under-fits the data. This is indicated by the fact that both the training and validation errors are very high.
Adding more data WON'T work in this case.
When both TRAINING and VALIDATION curves converge to an high error, we have a HIGH BIAS model. In this situation we can try one of the following actions:
- Using a more sophisticated model (i.e. in this case, increase
d) - Gather more features for each sample.
- Decrease regularlization.
Now let's look at a model with higher degree (and thus higher variance):
plot_learning_curve(d=10, N=100, y_range=(0,12))
In this example it is possible to see that with d=10 a high-variance estimator may overfit data when the number of samples is not sufficent. It is possible to note it by looking at the discrepancy between the two curves. When the number of samples is low we have a low training error, but the corresponding test error is really high. As the number of samples increase the test error decrease and converge to the model error but it may be to slow to converge and still have an higher discrepancy between the two curves.
When the learning curves does not converge using the full training set, it indicates a HIGH VARIANCE model, and the model is OVERFITTING the data.
A high-variance model can be improved by:
- Gathering more training samples.
- Using a less-sophisticated model (i.e. in this case, make
dsmaller) - Increase regularization.
In particular, gathering more features for each sample will not help the results.
6 Do the right thing¶
6.1 High Bias¶
If our algorithm shows high bias, the following actions might help:
- Add more features. In our example of predicting home prices, it may be helpful to make use of information such as the neighborhood the house is in, the year the house was built, the size of the lot, etc. Adding these features to the training and test sets can improve a high-bias estimator
- Use a more sophisticated model. Adding complexity to the model can help improve on bias. For a polynomial fit, this can be accomplished by increasing the degree d. Each learning technique has its own methods of adding complexity.
- Use fewer samples. Though this will not improve the classification, a high-bias algorithm can attain nearly the same error with a smaller training sample. For algorithms which are computationally expensive, reducing the training sample size can lead to very large improvements in speed.
- Decrease regularization. Regularization is a technique used to impose simplicity in some machine learning models, by adding a penalty term that depends on the characteristics of the parameters. If a model has high bias, decreasing the effect of regularization can lead to better results.
6.2 High Variance¶
If our algorithm shows high variance, the following actions might help:
- Use fewer features. Using a feature selection technique may be useful, and decrease the over-fitting of the estimator.
- Use a simpler model. Model complexity and over-fitting go hand-in-hand.
- Use more training samples. Adding training samples can reduce the effect of over-fitting, and lead to improvements in a high variance estimator.
- Increase Regularization. Regularization is designed to prevent over-fitting. In a high-variance model, increasing regularization can lead to better results.
These choices become very important in real-world situations. For example, due to limited telescope time, astronomers must seek a balance between observing a large number of objects, and observing a large number of features for each object. Determining which is more important for a particular learning task can inform the observing strategy that the astronomer employs. In a later exercise, we will explore the use of learning curves for the photometric redshift problem.
6.3 More Sophisticate Methods¶
There are a lot more options for performing validation and model testing. In particular, there are several schemes for cross-validation, in which the model is fit multiple times with different training and test sets. The details are different, but the principles are the same as what we've seen here.
For more information see the sklearn.model_selection module documentation,
and the information on the scikit-learn website.
6.4 One Last Caution¶
Using validation schemes to determine hyper-parameters means that we are fitting the hyper-parameters to the particular validation set. In the same way that parameters can be over-fit to the training set, hyperparameters can be over-fit to the validation set. Because of this, the validation error tends to under-predict the classification error of new data.
For this reason, it is recommended to split the data into three sets:
- The training set, used to train the model (usually ~60% of the data)
- The validation set, used to validate the model (usually ~20% of the data)
- The test set, used to evaluate the expected error of the validated model (usually ~20% of the data)
This may seem excessive, and many machine learning practitioners ignore the need for a test set. But if your goal is to predict the error of a model on unknown data, using a test set is vital.
7 Cross Validation (CV)¶
By partitioning the available data into Training and Validation sets, we reduce the number of samples which can be used for learning the model.
By using cross-validation (CV) the validation set is defined automatically by the CV algoritm. The training set is split into k smaller sets and:
- A model is trained using k-1 of the folds as training data
- The resulting model is validated on the remaining part of the data
Image('images/k-fold.png')

In this example we first calculate the Mean Squared Error on the Train DatasetSet (which is a methodological error) and then we calculate again the same MSE using a KFold CV. The MSE calculated with Cv is much larger:
X, y = datasets.samples_generator.make_regression(n_samples=7,
n_features=1, n_informative=1,
random_state=0, noise=5)
lin = linear_model.LinearRegression(fit_intercept=True)
lin.fit(X, y)
MSE = metrics.mean_squared_error(lin.predict(X), y)
print('MSE Calculated on the Training Set: ', MSE)
MSE_CV = model_selection.cross_val_score(lin, X, y, cv=5, scoring='neg_mean_squared_error')
print("MSE Calculated with Cross Validation: %0.2f (+/- %0.2f)"
% (abs(MSE_CV).mean(), abs(MSE_CV).std() * 2))
MSE Calculated on the Training Set: 19.58765452634401 MSE Calculated with Cross Validation: 49.85 (+/- 80.06)
When the cv argument is an integer like cv=10, cross_val_score uses the KFold or StratifiedKFold strategies. It is also possible to use other cross validation strategies by passing a cross validation iterator instead, for instance we can use model_selection.ShuffleSplit where n_iter is the number of re-shuffling and splitting operations:
n_samples = X.shape[0]
cv_iter = model_selection.ShuffleSplit(n_samples, test_size=0.2, random_state=0)
MSE_CV = model_selection.cross_val_score(lin, X, y, cv=cv_iter, scoring='neg_mean_squared_error')
print("MSE Calculated with Cross Validation: %0.2f (+/- %0.2f)"
% (abs(MSE_CV).mean(), abs(MSE_CV).std() * 2))
MSE Calculated with Cross Validation: 31.07 (+/- 36.74)
7.1 Cross Validation: test many estimators on the same dataset:¶
In this example we use the "diabetes" dataset to fit tree different linear models. For each model the default CV score is calculated and displayed:
data = datasets.load_diabetes()
X, y = data.data, data.target
for Model in [linear_model.LinearRegression,
linear_model.Ridge,
linear_model.Lasso]:
model = Model()
print(Model.__name__, ': ', model_selection.cross_val_score(model, X, y).mean())
LinearRegression : 0.4887027672471189 Ridge : 0.40942743830329875 Lasso : 0.35380008329932017
7.2 Cross Validation: test many hyperparamaters and estimators on the same dataset:¶
Lasso and Ridge accept a regularization parameter alpha. Here we plot the CV Score for different values of alpha
from bokeh.models.axes import LogAxis
alphas = np.logspace(-4, -1, 200)
fig = bk.figure(title='Alpha sensitiveness on different models',
plot_width=700, plot_height=300,
x_axis_type='log',
x_range=(min(alphas), max(alphas)))
for Model, color in [(linear_model.Ridge, 'blue'), (linear_model.Lasso, 'green')]:
scores = [model_selection.cross_val_score(Model(alpha=a), X, y, cv=5).mean()
for a in alphas]
fig.line(alphas, scores, line_color=color, legend=Model.__name__)
fig.legend.location = 'bottom_left'
bk.show(fig)
7.3 Model specific Cross Validation:¶
Some models can fit data for a range of parameters almost as efficiently as fitting for a single parameter. The most common parameter amenable to this strategy is the strength of the regularizer. In this case we say that we compute the regularization path of the estimator.
This is a particular case of what we are going to see in the next paragraph: Grid Search.
Model Specific Cross Validation is supported by the following models:
- RidgeCV
- RidgeClassifierCV
- LarsCV
- LassoLarsCV
- LassoCV
- ElasticNetCV
for Model in [linear_model.RidgeCV, linear_model.LassoCV]:
model = Model(alphas=alphas, cv=5).fit(X, y)
print(Model.__name__, ': ', model.alpha_)
RidgeCV : 0.0003739937302478798 LassoCV : 0.003827494478516311
7.4 Cross-validation iterators¶
- K-fold - divide all samples in K groups, if K=n it is equivalent to leave-one-out.
- Statified K-fold - each set contains the same prercentage of the target classes as the whole set
- Leave-One-Out (LOO) - all samples except one
- Leave-P-Out (LPO) - create all possible training sets by removing p samples from the whole set
- Leave-One-Label-Out (LOLO) - holds out the samples according to a third-party provided label
- ShuffleSplit - shuffles then split data in train and test sets
- StratifiedShuffleSplit - ShuffleSplits by preserving percentage
- Bootstrap - generate independent splits and then resample with replacement each split
X = np.arange(20).reshape(10,2)
y = np.arange(10)
KFold split dataset into k consecutive folds (without shuffling):
kf = model_selection.KFold(n_splits=5)
for train, test in kf.split(X):
print(train, test)
[2 3 4 5 6 7 8 9] [0 1] [0 1 4 5 6 7 8 9] [2 3] [0 1 2 3 6 7 8 9] [4 5] [0 1 2 3 4 5 8 9] [6 7] [0 1 2 3 4 5 6 7] [8 9]
ShuffleSplit do not guarantee that all folds will be different:
ss = model_selection.ShuffleSplit(n_splits=5, test_size=0.2)
for train, test in ss.split(X):
print(train, test)
[5 3 9 8 0 7 1 4] [6 2] [1 9 2 3 5 8 7 4] [0 6] [4 3 6 2 9 5 7 0] [8 1] [9 5 8 7 2 0 1 6] [4 3] [5 8 2 3 1 6 4 7] [9 0]
Bootstrap example:
bs = model_selection.LeaveOneOut()
for train, test in bs.split(X):
print(train, test)
[1 2 3 4 5 6 7 8 9] [0] [0 2 3 4 5 6 7 8 9] [1] [0 1 3 4 5 6 7 8 9] [2] [0 1 2 4 5 6 7 8 9] [3] [0 1 2 3 5 6 7 8 9] [4] [0 1 2 3 4 6 7 8 9] [5] [0 1 2 3 4 5 7 8 9] [6] [0 1 2 3 4 5 6 8 9] [7] [0 1 2 3 4 5 6 7 9] [8] [0 1 2 3 4 5 6 7 8] [9]
8 Grid Search: Searching for estimator hyperparameters¶
GridSearchCV can optimize any parameter provided by estimator.get_params() by exhaustively fitting the model with any parameter combination.
When evaluating different settings (“hyperparameters”) for estimators.
GridSearchCV will optimize the hyperparameters on the TRAINING set (whereas the VALIDATION set is generated by CV). After the optimization has been done, the performances of the estimator can be evaluated on a third set called TEST set:
8.1 Exhaustive Grid Search¶
We'll pass to GridSearchCV a dictionary of parameters containing a list of hyperparameters to be searched. In this case we set verbose=2 to see an output about all the searches done by the algorithm:
from sklearn import svm
# Standardize the data
iris = datasets.load_iris()
scaler = preprocessing.StandardScaler(copy=True, with_mean=True, with_std=True)
X = scaler.fit_transform(iris.data)
# Keep out the Test Set
X_tr, X_test, y_tr, y_test = model_selection.train_test_split(X, iris.target,
test_size=0.30, random_state=0)
# Define the Search Grid
param_grid = [{'C': [1, 10], 'kernel': ['linear']},
{'C': [2], 'gamma': [0.1, 0.01], 'kernel': ['rbf']}]
# GridSearchCV
clf = model_selection.GridSearchCV(svm.SVC(), param_grid, cv=5, n_jobs=-1, verbose=2)
clf.fit(X_tr, y_tr)
Fitting 5 folds for each of 4 candidates, totalling 20 fits [CV] C=1, kernel=linear .............................................. [CV] C=1, kernel=linear .............................................. [CV] C=1, kernel=linear .............................................. [CV] ............................... C=1, kernel=linear, total= 0.0s [CV] ............................... C=1, kernel=linear, total= 0.0s [CV] C=1, kernel=linear .............................................. [CV] C=10, kernel=linear ............................................. [CV] ............................... C=1, kernel=linear, total= 0.0s [CV] C=10, kernel=linear ............................................. [CV] C=10, kernel=linear ............................................. [CV] C=2, gamma=0.1, kernel=rbf ...................................... [CV] C=2, gamma=0.1, kernel=rbf ...................................... [CV] C=2, gamma=0.1, kernel=rbf ...................................... [CV] ............................... C=1, kernel=linear, total= 0.0s [CV] .............................. C=10, kernel=linear, total= 0.0s [CV] C=2, gamma=0.1, kernel=rbf ...................................... [CV] .............................. C=10, kernel=linear, total= 0.0s [CV] .............................. C=10, kernel=linear, total= 0.0s [CV] ....................... C=2, gamma=0.1, kernel=rbf, total= 0.0s [CV] C=2, gamma=0.01, kernel=rbf ..................................... [CV] ....................... C=2, gamma=0.1, kernel=rbf, total= 0.0s [CV] ....................... C=2, gamma=0.1, kernel=rbf, total= 0.0s [CV] C=2, gamma=0.1, kernel=rbf ...................................... [CV] C=2, gamma=0.01, kernel=rbf ..................................... [CV] C=2, gamma=0.01, kernel=rbf ..................................... [CV] C=2, gamma=0.01, kernel=rbf ..................................... [CV] C=2, gamma=0.01, kernel=rbf ..................................... [CV] ...................... C=2, gamma=0.01, kernel=rbf, total= 0.0s [CV] ...................... C=2, gamma=0.01, kernel=rbf, total= 0.0s [CV] ...................... C=2, gamma=0.01, kernel=rbf, total= 0.0s [CV] ....................... C=2, gamma=0.1, kernel=rbf, total= 0.0s [CV] ....................... C=2, gamma=0.1, kernel=rbf, total= 0.0s [CV] ...................... C=2, gamma=0.01, kernel=rbf, total= 0.0s [CV] ...................... C=2, gamma=0.01, kernel=rbf, total= 0.0s [CV] C=1, kernel=linear .............................................. [CV] ............................... C=1, kernel=linear, total= 0.0s [CV] C=10, kernel=linear ............................................. [CV] .............................. C=10, kernel=linear, total= 0.0s [CV] C=10, kernel=linear ............................................. [CV] .............................. C=10, kernel=linear, total= 0.0s
[Parallel(n_jobs=-1)]: Done 8 out of 20 | elapsed: 0.0s remaining: 0.0s [Parallel(n_jobs=-1)]: Done 20 out of 20 | elapsed: 0.0s finished
GridSearchCV(cv=5, error_score='raise',
estimator=SVC(C=1.0, cache_size=200, class_weight=None, coef0=0.0,
decision_function_shape='ovr', degree=3, gamma='auto', kernel='rbf',
max_iter=-1, probability=False, random_state=None, shrinking=True,
tol=0.001, verbose=False),
fit_params=None, iid=True, n_jobs=-1,
param_grid=[{'C': [1, 10], 'kernel': ['linear']}, {'C': [2], 'gamma': [0.1, 0.01], 'kernel': ['rbf']}],
pre_dispatch='2*n_jobs', refit=True, return_train_score='warn',
scoring=None, verbose=2)
for a,b,c in zip(clf.cv_results_['mean_test_score'],
clf.cv_results_['std_test_score'],
clf.cv_results_['params']):
print('mean: {:.3f}, std: {:.4f}, params: {}'.format(a,b,c))
mean: 0.943, std: 0.0348, params: {'C': 1, 'kernel': 'linear'}
mean: 0.933, std: 0.0469, params: {'C': 10, 'kernel': 'linear'}
mean: 0.943, std: 0.0348, params: {'C': 2, 'gamma': 0.1, 'kernel': 'rbf'}
mean: 0.895, std: 0.0981, params: {'C': 2, 'gamma': 0.01, 'kernel': 'rbf'}
Then we can check the accuracy on the Test Set:
best = clf.best_estimator_
print('Best estimator: ', best)
scores = model_selection.cross_val_score(best, X_test, y_test, cv=10)
print("Accuracy: %0.2f (+/- %0.2f)" % (scores.mean(), scores.std() * 2))
Best estimator: SVC(C=1, cache_size=200, class_weight=None, coef0=0.0, decision_function_shape='ovr', degree=3, gamma='auto', kernel='linear', max_iter=-1, probability=False, random_state=None, shrinking=True, tol=0.001, verbose=False) Accuracy: 0.91 (+/- 0.22)
Different score metrics can be used while doing GridSearchCV:
X_tr, X_test, y_tr, y_test = model_selection.train_test_split(X,
iris.target,
test_size=0.60,
random_state=0)
scores = ['precision_weighted', 'recall_weighted']
param_grid = [{'C': np.logspace(-3,0,4, base=2), 'kernel': ['linear']},
{'C': [1, 10], 'gamma': [0.1], 'kernel': ['rbf']}]
print('PRECISION: fraction of retrieved instances that are relevant')
print('RECALL: fraction of relevant instances that are retrieved')
for score in scores:
print('\nTuning hyper-parameters for: %s' % score)
clf = model_selection.GridSearchCV(svm.SVC(), param_grid, cv=5, scoring=score)
clf.fit(X_tr, y_tr)
print('\nBest parameters set found on development (training) set:')
print(clf.best_estimator_)
print('\nGrid Scores on development set:')
for a,b,c in zip(clf.cv_results_['mean_test_score'],
clf.cv_results_['std_test_score'],
clf.cv_results_['params']):
print('mean: {:.3f}, std: {:.4f}, params: {}'.format(a,b,c))
print('\nDetailed classification report (on Testing Set)')
y_true, y_pred = y_test, clf.predict(X_test)
print(metrics.classification_report(y_true, y_pred))
print('-----------------------------------------------------')
PRECISION: fraction of retrieved instances that are relevant
RECALL: fraction of relevant instances that are retrieved
Tuning hyper-parameters for: precision_weighted
Best parameters set found on development (training) set:
SVC(C=0.5, cache_size=200, class_weight=None, coef0=0.0,
decision_function_shape='ovr', degree=3, gamma='auto', kernel='linear',
max_iter=-1, probability=False, random_state=None, shrinking=True,
tol=0.001, verbose=False)
Grid Scores on development set:
mean: 0.920, std: 0.0815, params: {'C': 0.125, 'kernel': 'linear'}
mean: 0.953, std: 0.0648, params: {'C': 0.25, 'kernel': 'linear'}
mean: 0.987, std: 0.0267, params: {'C': 0.5, 'kernel': 'linear'}
mean: 0.974, std: 0.0340, params: {'C': 1.0, 'kernel': 'linear'}
mean: 0.941, std: 0.0617, params: {'C': 1, 'gamma': 0.1, 'kernel': 'rbf'}
mean: 0.974, std: 0.0340, params: {'C': 10, 'gamma': 0.1, 'kernel': 'rbf'}
Detailed classification report (on Testing Set)
precision recall f1-score support
0 1.00 1.00 1.00 26
1 0.89 1.00 0.94 33
2 1.00 0.87 0.93 31
avg / total 0.96 0.96 0.96 90
-----------------------------------------------------
Tuning hyper-parameters for: recall_weighted
Best parameters set found on development (training) set:
SVC(C=0.5, cache_size=200, class_weight=None, coef0=0.0,
decision_function_shape='ovr', degree=3, gamma='auto', kernel='linear',
max_iter=-1, probability=False, random_state=None, shrinking=True,
tol=0.001, verbose=False)
Grid Scores on development set:
mean: 0.917, std: 0.0810, params: {'C': 0.125, 'kernel': 'linear'}
mean: 0.950, std: 0.0659, params: {'C': 0.25, 'kernel': 'linear'}
mean: 0.983, std: 0.0333, params: {'C': 0.5, 'kernel': 'linear'}
mean: 0.967, std: 0.0441, params: {'C': 1.0, 'kernel': 'linear'}
mean: 0.933, std: 0.0637, params: {'C': 1, 'gamma': 0.1, 'kernel': 'rbf'}
mean: 0.967, std: 0.0441, params: {'C': 10, 'gamma': 0.1, 'kernel': 'rbf'}
Detailed classification report (on Testing Set)
precision recall f1-score support
0 1.00 1.00 1.00 26
1 0.89 1.00 0.94 33
2 1.00 0.87 0.93 31
avg / total 0.96 0.96 0.96 90
-----------------------------------------------------
8.2 Randomized Parameter Optimization¶
RandomizedSearchCV implements a randomized search over parameters, where each setting is sampled from a distribution over possible parameter values. This has two main benefits:
- A budget can be chosen independent of the number of parameters and possible values.
- Adding parameters that do not influence the performance does not decrease efficiency.
Specifying how parameters should be sampled is done using a dictionary. The number of sampling iterations, is specified using the n_iter parameter. For each parameter, either a distribution over possible values or a list of discrete choices (which will be sampled uniformly) can be specified. This is a little example of the many statistical distributions available in scipy:
import scipy.stats
w, h = 350, 300
dists = [
scipy.stats.expon(scale=100), # the frozed uniform distribution
scipy.stats.norm(loc=40, scale=10), # the frozed uniform distribution
scipy.stats.uniform(loc=20, scale=100), # the frozed uniform distribution
scipy.stats.beta(2,2,loc=20, scale=100) # the frozed uniform distribution
]
figs = []
for dist in dists:
hist, edges = np.histogram(dist.rvs(int(1e4)), density=True, bins=50)
p = bk.figure(plot_width=w, plot_height=h)
p.quad(top=hist, bottom=0, left=edges[:-1], right=edges[1:])
figs.append(p)
bk.show(bk.gridplot([[figs[0], figs[1]],
[figs[2], figs[3]]
]))
# Standardize the data
iris = datasets.load_iris()
scaler = preprocessing.StandardScaler(copy=True, with_mean=True, with_std=True)
X = scaler.fit_transform(iris.data)
# Keep out the Test Set
X_tr, X_test, y_tr, y_test = model_selection.train_test_split(X, iris.target,
test_size=0.30, random_state=0)
# Define the Search Grid
param_grid = {'C': scipy.stats.uniform(loc=1, scale=100), 'kernel': ['linear','rbf'],
'gamma': scipy.stats.uniform(loc=0.01, scale=10)}
# GridSearchCV
clf = model_selection.RandomizedSearchCV(svm.SVC(), param_grid, cv=5, n_jobs=-1, verbose=2, n_iter=10)
clf.fit(X_tr, y_tr)
Fitting 5 folds for each of 10 candidates, totalling 50 fits [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear .... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear .... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear .... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear, total= 0.0s [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear ..... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear, total= 0.0s [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear ... [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear, total= 0.0s [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear ... [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear ..... [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear ..... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear, total= 0.0s [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear, total= 0.0s [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear, total= 0.0s [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear ... [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear, total= 0.0s [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear ... [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear .... [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear ... [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear, total= 0.0s [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear .... [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear, total= 0.0s [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear, total= 0.0s [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear, total= 0.0s [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear, total= 0.0s [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear ..... [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear .... [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear .... [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf ......... [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf ......... [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear, total= 0.0s [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf ......... [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear .... [CV] C=17.155859565449827, gamma=1.6913486247050757, kernel=linear, total= 0.0s [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf ......... [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf, total= 0.0s [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear, total= 0.0s [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear ..... [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf ......... [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear, total= 0.0s [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf ....... [CV] C=93.31554616176741, gamma=1.3888061075606983, kernel=linear, total= 0.0s [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf, total= 0.0s [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf, total= 0.0s [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf, total= 0.0s [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf, total= 0.0s [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear .... [CV] C=55.93446930891519, gamma=3.894385938747857, kernel=linear, total= 0.0s [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear ..... [CV] C=48.06153787597535, gamma=8.83503861242045, kernel=rbf, total= 0.0s [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf ....... [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear, total= 0.0s [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear ..... [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf, total= 0.0s [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear, total= 0.0s [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear ..... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear, total= 0.0s [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear, total= 0.0s [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear ..... [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear, total= 0.0s [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear ..... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear .... [CV] C=66.95572367936933, gamma=9.537084781812451, kernel=linear, total= 0.0s [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf ......... [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf, total= 0.0s [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf ......... [CV] C=26.414479945822865, gamma=8.461920981407586, kernel=linear, total= 0.0s [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf, total= 0.0s [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf ......... [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf, total= 0.0s [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf ......... [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf, total= 0.0s [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf ......... [CV] C=83.3401698120732, gamma=6.423318313980214, kernel=rbf, total= 0.0s [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf .......... [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf, total= 0.0s [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf .......... [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf, total= 0.0s [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf .......... [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf, total= 0.0s [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf .......... [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf, total= 0.0s [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf .......... [CV] C=68.48252946299827, gamma=9.5154416062423, kernel=rbf, total= 0.0s [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear ...... [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear, total= 0.0s [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear ...... [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear, total= 0.0s [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear ...... [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear, total= 0.0s [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear ...... [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear, total= 0.0s [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear ...... [CV] C=60.95882351047934, gamma=8.31572018382552, kernel=linear, total= 0.0s [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf ....... [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf, total= 0.0s [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf ....... [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf, total= 0.0s [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf ....... [CV] C=3.4384495595874522, gamma=5.027487326691249, kernel=rbf, total= 0.0s
[Parallel(n_jobs=-1)]: Done 50 out of 50 | elapsed: 0.1s finished
RandomizedSearchCV(cv=5, error_score='raise',
estimator=SVC(C=1.0, cache_size=200, class_weight=None, coef0=0.0,
decision_function_shape='ovr', degree=3, gamma='auto', kernel='rbf',
max_iter=-1, probability=False, random_state=None, shrinking=True,
tol=0.001, verbose=False),
fit_params=None, iid=True, n_iter=10, n_jobs=-1,
param_distributions={'C': <scipy.stats._distn_infrastructure.rv_frozen object at 0x7fb2c353b4e0>, 'kernel': ['linear', 'rbf'], 'gamma': <scipy.stats._distn_infrastructure.rv_frozen object at 0x7fb2c353b5f8>},
pre_dispatch='2*n_jobs', random_state=None, refit=True,
return_train_score='warn', scoring=None, verbose=2)
for a,b,c in zip(clf.cv_results_['mean_test_score'],
clf.cv_results_['std_test_score'],
clf.cv_results_['params']):
print('mean: {:.3f}, std: {:.4f}, params: {}'.format(a,b,c))
mean: 0.962, std: 0.0360, params: {'C': 26.414479945822865, 'gamma': 8.461920981407586, 'kernel': 'linear'}
mean: 0.962, std: 0.0360, params: {'C': 55.93446930891519, 'gamma': 3.894385938747857, 'kernel': 'linear'}
mean: 0.943, std: 0.0348, params: {'C': 17.155859565449827, 'gamma': 1.6913486247050757, 'kernel': 'linear'}
mean: 0.962, std: 0.0360, params: {'C': 93.31554616176741, 'gamma': 1.3888061075606983, 'kernel': 'linear'}
mean: 0.857, std: 0.0813, params: {'C': 48.06153787597535, 'gamma': 8.83503861242045, 'kernel': 'rbf'}
mean: 0.962, std: 0.0360, params: {'C': 66.95572367936933, 'gamma': 9.537084781812451, 'kernel': 'linear'}
mean: 0.895, std: 0.0676, params: {'C': 83.3401698120732, 'gamma': 6.423318313980214, 'kernel': 'rbf'}
mean: 0.857, std: 0.0813, params: {'C': 68.48252946299827, 'gamma': 9.5154416062423, 'kernel': 'rbf'}
mean: 0.962, std: 0.0360, params: {'C': 60.95882351047934, 'gamma': 8.31572018382552, 'kernel': 'linear'}
mean: 0.895, std: 0.0738, params: {'C': 3.4384495595874522, 'gamma': 5.027487326691249, 'kernel': 'rbf'}
best = clf.best_estimator_
print('Best estimator: ', best)
scores = model_selection.cross_val_score(best, X_test, y_test, cv=10)
print("Accuracy: %0.2f (+/- %0.2f)" % (scores.mean(), scores.std() * 2))
Best estimator: SVC(C=26.414479945822865, cache_size=200, class_weight=None, coef0=0.0, decision_function_shape='ovr', degree=3, gamma=8.461920981407586, kernel='linear', max_iter=-1, probability=False, random_state=None, shrinking=True, tol=0.001, verbose=False) Accuracy: 0.96 (+/- 0.18)
9 Pipelines¶
When we applied different preprocessing techniques, such as standardization for feature scaling or principal component analysis you learned that we have to reuse the parameters that were obtained during the fitting of the training data to scale and compress any new data, for example, the samples in the separate test dataset.
In this section, you will learn about an extremely handy tool, the Pipeline class in scikit-learn. It allows us to fit a model including an arbitrary number of transformation steps and apply it to make predictions about new data.
In this chapter, we will be working with the Breast Cancer Wisconsin dataset, which contains 569 samples of malignant and benign tumor cells. The first two columns in the dataset store the unique ID numbers of the samples and the corresponding diagnosis (M=malignant, B=benign), respectively. The columns 3-32 contain 30 real-value features that have been computed from digitized images of the cell nuclei, which can be used to build a model to predict whether a tumor is benign or malignant. The Breast Cancer Wisconsin dataset has been deposited on the UCI machine learning repository and more detailed information about this dataset can be found at https://archive.ics.uci.edu/ml/datasets/Breast+Cancer+Wisconsin+(Diagnostic)
In this section we will read in the dataset, and split it into training and test datasets in three simple steps.
- We will start by reading in the dataset directly from the UCI website using pandas:
import pandas as pd
df = pd.read_csv('https://archive.ics.uci.edu/ml/machine-learning-databases/breast-cancer-wisconsin/wdbc.data',
header=None)
- Next, we assign the 30 features to a NumPy array X . Using LabelEncoder, we transform the class labels from their original string representation ( M and B ) into integers:
from sklearn.preprocessing import LabelEncoder
X = df.loc[:, 2:].values
y = df.loc[:, 1].values
le = LabelEncoder()
y = le.fit_transform(y)
After encoding the class labels (diagnosis) in an array y , the malignant tumors are now represented as class 1 , and the benign tumors are represented as class 0 , respectively, which we can illustrate by calling the transform method of LabelEncoder on two dummy class labels:
le.transform(['M', 'B'])
array([1, 0])
- Before we construct our first model pipeline in the following subsection, let's divide the dataset into a separate training dataset (80 percent of the data) and a separate test dataset (20 percent of the data):
from sklearn.model_selection import train_test_split
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.20, random_state=1)
Many learning algorithms require input features on the same scale for optimal performance. Thus, we need to standardize the columns in the Breast Cancer Wisconsin dataset before we can feed them to a linear classifier, such as logistic regression. Furthermore, let's assume that we want to compress our data from the initial 30 dimensions onto a lower two-dimensional subspace via principal component analysis (PCA). Instead of going through the fitting and transformation steps for the training and test dataset separately, we can chain the StandardScaler, PCA , and LogisticRegression objects in a pipeline:
from sklearn.preprocessing import StandardScaler
from sklearn.decomposition import PCA
from sklearn.linear_model import LogisticRegression
from sklearn.pipeline import Pipeline
pipe_lr = Pipeline([('scl', StandardScaler()), ('pca', PCA(n_components=2)),
('clf', LogisticRegression(random_state=1))])
pipe_lr.fit(X_train, y_train)
print('Test Accuracy: %.3f' % pipe_lr.score(X_test, y_test))
Test Accuracy: 0.947
The intermediate steps in a pipeline constitute scikit-learn transformers, and the last step is an estimator. In the preceding code example, we built a pipeline that consisted of two intermediate steps, a StandardScaler and a PCA transformer, and a logistic regression classifier as a final estimator. When we executed the fit method on the pipeline pipe_lr , the StandardScaler performed fit and transform on the training data, and the transformed training data was then passed onto the next object in the pipeline, the PCA. Similar to the previous step, PCA also executed fit and transform on the scaled input data and passed it to the final element of the pipeline, the estimator. We should note that there is no limit to the number of intermediate steps in this pipeline.
10 Performance Metrics¶
In the previous sections and chapters, we evaluated our models using the mean squared error, which is a useful metric to quantify the performance of a model in general. However, there are several other performance metrics that can be used to measure a model's relevance, such as precision, recall, and the F1-score.
Before we get into the details of different scoring metrics, let's print a so-called confusion matrix, a matrix that lays out the performance of a learning algorithm. The confusion matrix is simply a square matrix that reports the counts of the true positive, true negative, false positive, and false negative predictions of a classifier.
Although these metrics can be easily computed manually by comparing the true and predicted class labels, cikit-learn provides a convenient confusion_matrix function that we can use as follows:
from sklearn.metrics import confusion_matrix
y_pred = pipe_lr.predict(X_test)
confmat = confusion_matrix(y_true=y_test, y_pred=y_pred)
print(confmat)
[[71 1] [ 5 37]]
The array that was returned after executing the preceding code provides us with information about the different types of errors the classifier made on the test dataset that we can map onto the confusion matrix illustration in the previous figure using matplotlib's matshow function:
fig, ax = plt.subplots(figsize=(5.5, 5.5))
ax.matshow(confmat, cmap=plt.cm.Blues, alpha=0.3)
for i in range(confmat.shape[0]):
for j in range(confmat.shape[1]):
ax.text(x=j, y=i,
s=confmat[i, j],
va='center', ha='center')
plt.xlabel('predicted label')
plt.ylabel('true label')
plt.show()

Assuming that class 1 (malignant) is the positive class in this example, our model correctly classified 71 of the samples that belong to class 0 (false negatives) and 37 samples that belong to class 1 (true positives), respectively. However, our model also incorrectly misclassified 5 samples from class 0 as class 1 (false negatives), and it predicted that 1 sample is benign although it is a malignant tumor (false positive). In the next section, we will learn how we can use this information to calculate various different error metrics.
Both the prediction error (ERR) and accuracy (ACC) provide general information about how many samples are misclassified. The error can be understood as the sum of all false predictions divided by the number of total predictions, and the accuracy is calculated as the sum of correct predictions divided by the total number of predictions, respectively:
$$ERR = \frac{FP + FN}{Total}$$The prediction accuracy can then be calculated directly from the error:
$$ACC = \frac{TP + TN}{Total} = 1 − ERR$$In tumor diagnosis, for example, we are more concerned about the detection of malignant tumors in order to help a patient with the appropriate treatment. However, it is also important to decrease the number of benign tumors that were incorrectly classified as malignant (false positives) to not unnecessarily concern a patient.
Precision (P) is defined as the number of true positives ($T_p$) over the number of true positives plus the number of false positives ($F_p$).
$$P = \frac{T_p}{T_p+F_p}$$Recall (R) is defined as the number of true positives ($T_p$) over the number of true positives plus the number of false negatives ($F_n$).
$$R = \frac{T_p}{T_p + F_n}$$These quantities are also related to the ($F_1$) score, which is defined as the harmonic mean of precision and recall.
$$F_1 = 2\frac{P \times R}{P+R}$$It is important to note that the precision may not decrease with recall. The definition of precision ($\frac{T_p}{T_p + F_p}$) shows that lowering the threshold of a classifier may increase the denominator, by increasing the number of results returned. If the threshold was previously set too high, the new results may all be true positives, which will increase precision. If the previous threshold was about right or too low, further lowering the threshold will introduce false positives, decreasing precision.
Recall is defined as $\frac{T_p}{T_p+F_n}$, where $T_p+F_n$ does not depend on the classifier threshold. This means that lowering the classifier threshold may increase recall, by increasing the number of true positive results. It is also possible that lowering the threshold may leave recall unchanged, while the precision fluctuates.
These scoring metrics are all implemented in scikit-learn and can be imported from the sklearn.metrics module:
from sklearn.metrics import precision_score
from sklearn.metrics import recall_score, f1_score
print('Precision: %.3f' % precision_score(y_true=y_test, y_pred=y_pred))
print('Recall: %.3f' % recall_score(y_true=y_test, y_pred=y_pred))
print('F1: %.3f' % f1_score(y_true=y_test, y_pred=y_pred))
Precision: 0.974 Recall: 0.881 F1: 0.925
Visit www.add-for.com for more tutorials and updates.
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.