** DIscBIO: a user-friendly pipeline for biomarker discovery in single-cell transcriptomics**¶
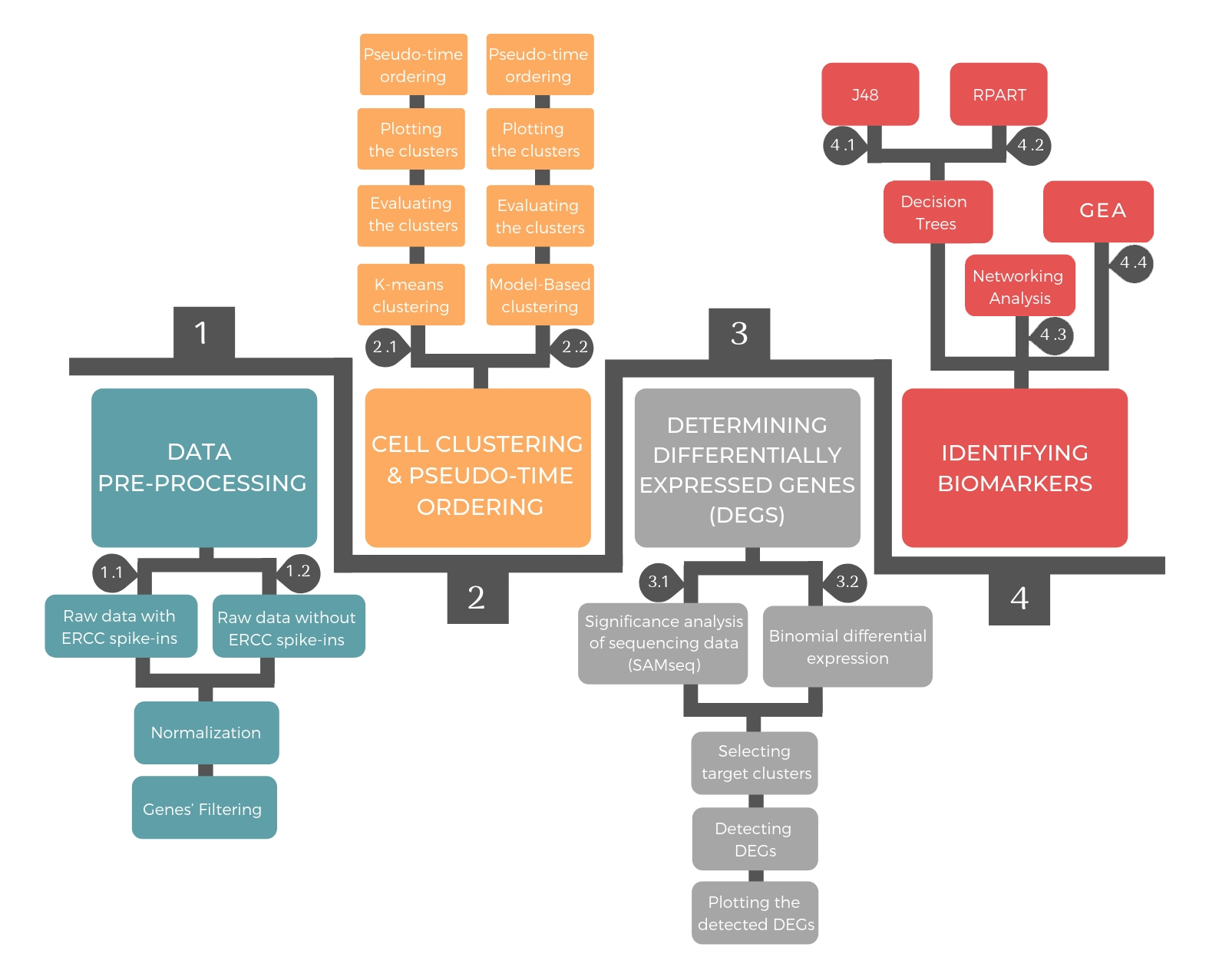
The pipeline consists of four successive steps: data pre-processing, cellular clustering and pseudo-temporal ordering, determining differential expressed genes and identifying biomarkers.
Required Packages¶
library(DIscBIO)
Loading required package: SingleCellExperiment
Loading required package: SummarizedExperiment
Loading required package: MatrixGenerics
Loading required package: matrixStats
Attaching package: ‘MatrixGenerics’
The following objects are masked from ‘package:matrixStats’:
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Loading required package: GenomicRanges
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which.max, which.min
Loading required package: S4Vectors
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:base’:
expand.grid
Loading required package: IRanges
Loading required package: GenomeInfoDb
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: ‘Biobase’
The following object is masked from ‘package:MatrixGenerics’:
rowMedians
The following objects are masked from ‘package:matrixStats’:
anyMissing, rowMedians
Loading dataset¶
The "CTCdataset" dataset consisting of single migratory circulating tumor cells (CTCs) collected from patients with breast cancer. Data are available in the GEO database with accession numbers GSE51827, GSE55807, GSE67939, GSE75367, GSE109761, GSE111065 and GSE86978. The dataset should be formatted in a data frame where columns refer to samples and rows refer to genes. We provide here the possibility to load the dataset either as ".csv" or ".rda" extensions.The dataset should be formatted in a data frame where columns refer to samples and rows refer to genes. We provide here the possibility to load the dataset either as ".csv" or ".rda" extensions.
FileName<-"CTCdataset" # Name of the dataset
#CSV=TRUE # If the dataset has ".csv", the user shoud set CSV to TRUE
CSV=FALSE # If the dataset has ".rda", the user shoud set CSV to FALSE
if (CSV==TRUE){
DataSet <- read.csv(file = paste0(FileName,".csv"), sep = ",",header=T)
rownames(DataSet)<-DataSet[,1]
DataSet<-DataSet[,-1]
} else{
load(paste0(FileName,".rda"))
DataSet<-get(FileName)
}
cat(paste0("The ", FileName," contains:","\n","Genes: ",length(DataSet[,1]),"\n","cells: ",length(DataSet[1,]),"\n"))
The CTCdataset contains: Genes: 13181 cells: 1462
1. Preparing the dataset¶
FG<- DISCBIO(DataSet)
FG<-Normalizedata(FG, mintotal=1000, minexpr=0, minnumber=0, maxexpr=Inf, downsample=FALSE, dsn=1, rseed=17000)
FG<-FinalPreprocessing(FG,GeneFlitering="ExpF",export = TRUE) # The GeneFiltering should be set to "ExpF"
GolgiFragGeneList<- read.csv(file = "GolgiFragGeneList.csv", sep = ",",header=F)
Data<-FG@fdata
genes<-rownames(Data)
gene_list<- GolgiFragGeneList[,1]
idx_genes <- is.element(genes,gene_list)
OAdf<-Data[idx_genes,]
FG@fdata<-OAdf
dim(FG@fdata)
cat(paste0("A list of ", length(OAdf[,1]), " genes will be used for the clustering","\n"))
The gene filtering method = Noise filtering The Filtered Normalized dataset contains: Genes: 13181 cells: 1448 The Filtered Normalized dataset was saved as: filteredDataset.Rdata
- 97
- 1448
A list of 97 genes will be used for the clustering
2. Cellular Clustering and Pseudo Time ordering¶
load("fg.RData") # Loading the "fg" object that includes the data of the k-means clustering
FG<-fg # Storing the data of fg in the FG
########## Removing the unneeded objects
rm(DataSet)
rm(fg)
rm(Data)
rm(OAdf)
plotGap(FG) ### Plotting gap statistics

############ Plotting the clusters
withr::with_options(repr.plot.width=12, repr.plot.height=12)
plottSNE(FG)

FG<-pseudoTimeOrdering(FG,quiet = TRUE, export = FALSE)
plotOrderTsne(FG)

Evaluating the stability and consistancy of the clusters¶
# Silhouette plot
withr::with_options(repr.plot.width=12, repr.plot.height=25)
plotSilhouette(FG,K=4) # K is the number of clusters

# Jaccard Plot
withr::with_options(repr.plot.width=12, repr.plot.height=12)
Jaccard(FG,Clustering="K-means", K=4, plot = TRUE) # Jaccard
- 0.749
- 0.671
- 0.545
- 0.772

Plotting the gene expression of a particular gene in a tSNE map¶
g='ENSG00000171611' #### Plotting the log expression of PTCRA
plotExptSNE(FG,g)

g='ENSG00000111057' #### Plotting the log expression of KRT18
plotExptSNE(FG,g)
