** DIscBIO: a user-friendly pipeline for biomarker discovery in single-cell transcriptomics**¶
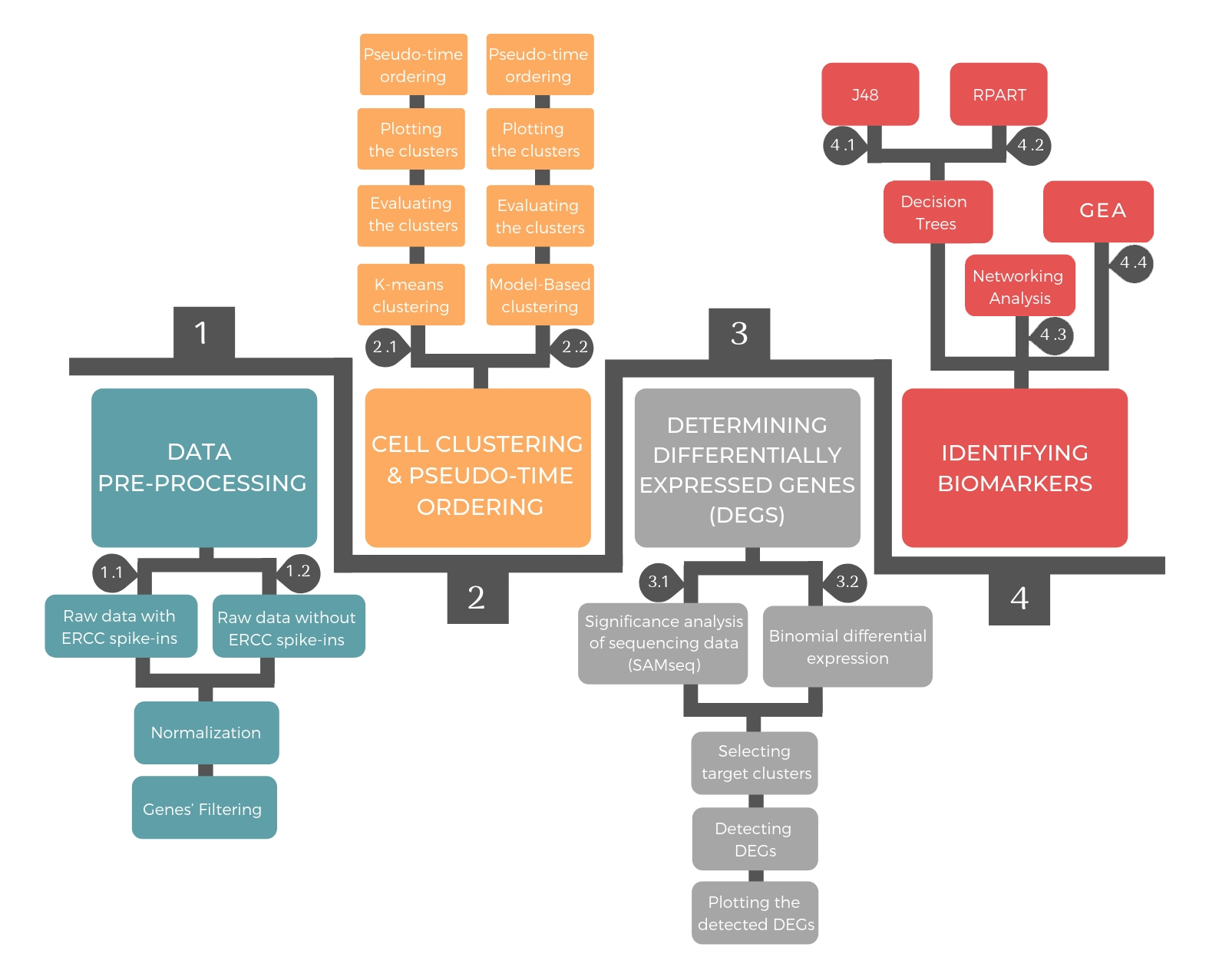
The pipeline consists of four successive steps: data pre-processing, cellular clustering and pseudo-temporal ordering, determining differential expressed genes and identifying biomarkers.
Required Packages¶
library(DIscBIO)
library(partykit)
library(enrichR)
Loading required package: SingleCellExperiment
Loading required package: SummarizedExperiment
Loading required package: MatrixGenerics
Loading required package: matrixStats
Attaching package: ‘MatrixGenerics’
The following objects are masked from ‘package:matrixStats’:
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Loading required package: GenomicRanges
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which.max, which.min
Loading required package: S4Vectors
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:base’:
expand.grid
Loading required package: IRanges
Loading required package: GenomeInfoDb
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: ‘Biobase’
The following object is masked from ‘package:MatrixGenerics’:
rowMedians
The following objects are masked from ‘package:matrixStats’:
anyMissing, rowMedians
Loading required package: grid
Loading required package: libcoin
Loading required package: mvtnorm
Attaching package: ‘partykit’
The following object is masked from ‘package:SummarizedExperiment’:
width
The following object is masked from ‘package:GenomicRanges’:
width
The following object is masked from ‘package:IRanges’:
width
The following object is masked from ‘package:S4Vectors’:
width
The following object is masked from ‘package:BiocGenerics’:
width
Welcome to enrichR
Checking connection ...
Connection is Live!
Loading dataset¶
The valuesG1ms dataset consists of single cells from a myxoid liposarcoma cell line. Myxoid liposarcoma is a rare type of tumor driven by specific fusion oncogenes, normally FUS-DDIT3 [Fletcher et al., 2013], with few other genetic changes [Ståhlberg et al., 2014; Hofvander et al., 2018]. The cells were collected based on their cell cycle phase (G1, S or G2/M), assessed by analyzing their DNA content using Fluorescence Activated Cell Sorter [Karlsson et al., 2017]. Data including ERCC spike-ins, are available in the ArrayExpress database at EMBL-EBI with accession number E-MTAB-6142).
The dataset should be formatted in a data frame where columns refer to samples and rows refer to genes. We provide here the possibility to load the dataset either as ".csv" or ".rda" extensions.
FileName<-"valuesG1ms" # Name of the dataset
CSV=TRUE # If the dataset has ".csv", the user shoud set CSV to TRUE
#CSV=FALSE # If the dataset has ".rda", the user shoud set CSV to FALSE
if (CSV==TRUE){
DataSet <- read.csv(file = paste0(FileName,".csv"), sep = ";",header=T)
rownames(DataSet)<-DataSet[,1]
DataSet<-DataSet[,-1]
} else{
load(paste0(FileName,".rda"))
DataSet<-get(FileName)
}
cat(paste0("The ", FileName," contains:","\n","Genes: ",length(DataSet[,1]),"\n","cells: ",length(DataSet[1,]),"\n"))
The valuesG1ms contains: Genes: 59838 cells: 94
sc<- DISCBIO(DataSet) # The DISCBIO class is the central object storing all information generated throughout the pipeline
1. Data Pre-processing¶
Prior to applying data analysis methods, it is standard to pre-process the raw read counts resulted from the sequencing. The preprocessing approach depends on the existence or absence of ERCC spike-ins. In both cases, it includes normalization of read counts and gene filtering.
Normalization of read counts¶
To account for RNA composition and sequencing depth among samples (single-cells), the normalization method “median of ratios” is used. This method takes the ratio of the gene instantaneous median to the total counts for all genes in that cell (column median). The gene instantaneous median is the product of multiplying the median of the total counts across all cells (row median) with the read of the target gene in each cell. This normalization method makes it possible to compare the normalized counts for each gene equally between samples.
Gene filtering¶
The key idea in filtering genes is to appoint the genes that manifest abundant variation across samples. Filtering genes is a critical step due to its dramatic impact on the downstream analysis. In case the raw data includes ERCC spike-ins, genes will be filtered based on variability in comparison to a noise level estimated from the ERCC spike-ins according to an algorithm developed by Brennecke et al (Brennecke et al., 2013). This algorithm utilizes the dependence of technical noise on the average read count and fits a model to the ERCC spike-ins. Further gene filtering can be implemented based on gene expression. In case the raw data does not include ERCC spike-ins, genes will be only filtered based on minimum expression in certain number of cells.

1.1. Filtering and normalizing the raw data that includes ERCCs¶
Filtering the raw data that includes ERCCs can be done by applying the “NoiseFiltering” function, which includes several parameters:
- object: the outcome of running the DISCBIO() function.
- percentile: A numeric value of the percentile. It is used to validate the ERCC spik-ins. Default is 0.8.
- CV: A numeric value of the coefficient of variation. It is used to validate the ERCC spik-ins. Default is 0.5.
- geneCol: Color of the genes that did not pass the filtration.
- FgeneCol: Color of the genes that passt the filtration.
- erccCol: Color of the ERCC spik-ins.
- Val: A logical vector that allows plotting only the validated ERCC spike-ins. Default is TRUE. If Val=FALSE will
plot all the ERCC spike-ins.
- plot: A logical vector that allows plotting the technical noise. Default is TRUE.
- export: A logical vector that allows writing the final gene list in excel file. Default is TRUE.
- quiet: if TRUE, suppresses printed output
To normalize the raw sequencing reads the function Normalizedata() should be used, this function takes 8 parameters.
- In case the user would like just to normalize the reads without any further gene filtering the parameters minexpr and minnumber should be set to 0.
- In case the user would like just to normalize the reads and run gene filtering based on gene expression the parameters minexpr and minnumber should have values. This function will discard cells with less than mintotal transcripts. Genes that are not expressed at minexpr transcripts in at least minnumber cells are discarded.
The function Normalizedata() normalizes the count reads using the normalization method “median of ratios”
To Finalize the preprocessing the function FinalPreprocessing() should be implemented by setting the parameter "GeneFlitering" to NoiseF ( whether the dditional gene filtering step based on gene expression was implemented on not).
sc<-NoiseFiltering(sc,percentile=0.8, CV=0.3)
#### Normalizing the reads without any further gene filtering
sc<-Normalizedata(sc, mintotal=1000, minexpr=0, minnumber=0, maxexpr=Inf, downsample=FALSE, dsn=1, rseed=17000)
sc<-FinalPreprocessing(sc,GeneFlitering="NoiseF",export = TRUE) ### The GeneFiltering can be either "NoiseF" or"ExpF"
Cut-off value for the ERCCs= 12.5 Coefficients of the fit:
a0 a1tilde 0.0200366 70.4893629
Explained variances of log CV^2 values= 0.84 Number of genes that passed the filtering = 5684 The gene filtering method = Noise filtering The Filtered Normalized dataset contains: Genes: 5684 cells: 94 The Filtered Normalized dataset was saved as: filteredDataset.Rdata

2. Cellular Clustering and Pseudo Time ordering¶
Cellular clustering is performed according to the gene expression profiles to detect cellular sub-population with unique properties. After clustering, pseudo-temporal ordering is generated to indicate the cellular differentiation degree.
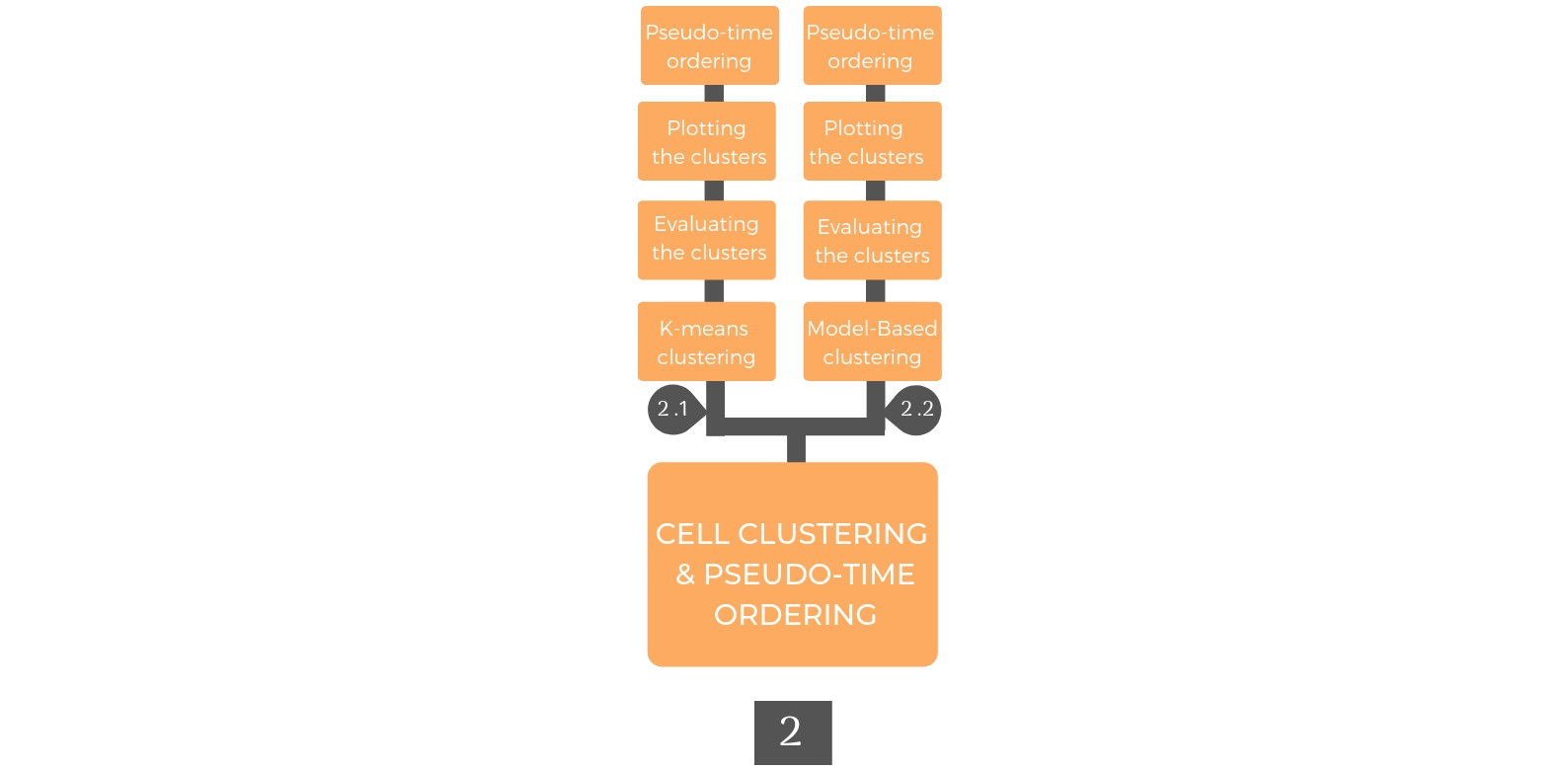
2.2. Model-Based clustering¶
Model-based clustering assumes that the data are generated by a model and attempts to recover the original model from the data to define cellular clusters.
2.2.1. Defining the Cells in the clusters generated by model-based clustering¶
sc <- Exprmclust(sc,K =3,reduce = TRUE,quiet = TRUE)
2.2.2. Cluster plotting using PCA and tSNE maps¶
To visualize the detected clusters, two common dimensionality reduction tools are implemented: tSNE map and principal component analysis (PCA), which is a linear dimensionality reduction method that preserves the global structure and shows how the measurements themselves are related to each other.
PlotmclustMB(sc)
PCAplotSymbols(sc)


# Plotting the model-based clusters in tSNE maps
sc<- comptSNE(sc,rseed=15555,quiet = F) # to perform the computation of a t-SNE map
plottSNE(sc)
This function may take time sigma summary: Min. : 38.4914978648748 |1st Qu. : 42.9672869120877 |Median : 45.7336241172451 |Mean : 47.2423249643292 |3rd Qu. : 50.7105126779753 |Max. : 73.7307741725683 | Epoch: Iteration #500 error is: 0.608233282504434 Epoch: Iteration #1000 error is: 0.597577895108978 Epoch: Iteration #1500 error is: 0.597054005007593 Epoch: Iteration #2000 error is: 0.59478833216114 Epoch: Iteration #2500 error is: 0.785133695910099 Epoch: Iteration #3000 error is: 0.467442752493165 Epoch: Iteration #3500 error is: 0.467349316975901 Epoch: Iteration #4000 error is: 0.467341397783741 Epoch: Iteration #4500 error is: 0.467329835708217 Epoch: Iteration #5000 error is: 0.467315499731271

2.2.3. Evaluating the stability and consistancy of the clusters¶
# Silhouette of Model-based clusters
withr::with_par(mar=c(6,2,4,2))
plotSilhouette(sc,K=3)

Jaccard(sc,Clustering="MB", K=3, plot = TRUE) # Jaccard of Model based clusters
- 0.66
- 0.766
- 0.812

Defining outlier cells based on Model-Based Clustering¶
Outlier identification is implemented using a background model based on distribution of transcript counts within a cluster. The background model is computed using the mean and the variance of the expression of each gene in a cluster. Outliers are defined as cells with a minimum of “outlg” outlier genes. Here we are setting the minimum number of outlier genes (the “outlg” parameter) to 5% of the number of filtered genes, this is based on the recommendation of De Vienne et al. (De Vienne et al., 2012).
In case the user decided to remove outlier cells, the user should set RemovingOutliers to TRUE and then start from the beginning (Data Pre-processing).
outlg<-round(length(sc@fdata[,1]) * 0.05) # The cell will be considered as an outlier if it has a minimum of 0.5% of the number of filtered genes as outlier genes.
Outliers<- FindOutliers(sc, K=3, outminc=5,outlg=outlg,probthr=.5*1e-3,thr=2**-(1:40),outdistquant=.75, plot = TRUE, quiet = T)
RemovingOutliers=FALSE
# RemovingOutliers=TRUE # Removing the defined outlier cells based on K-means Clustering
if(RemovingOutliers==TRUE){
names(Outliers)=NULL
Outliers
DataSet=DataSet[-Outliers]
dim(DataSet)
colnames(DataSet)
cat("Outlier cells were removed, now you need to start from the beginning")
}

2.2.4. Cellular pseudo-time ordering based on Model-based clusters¶
sc<-pseudoTimeOrdering(sc,quiet = FALSE, export = FALSE)
order orderID 1 1 G2_20 2 2 G2_17 3 3 G2_14 4 4 G2_13 5 5 G2_19 6 6 G1_25 7 7 G2_4 8 8 G2_18 9 9 G2_22 10 10 G2_23 11 11 G2_5 12 12 G2_7 13 13 G2_28 14 14 G2_25 15 15 G2_6 16 16 G2_27 17 17 G2_1 18 18 G1_16 19 19 G2 20 20 G2_10 21 21 G2_12 22 22 G2_29 23 23 G1_7 24 24 G2_24 25 25 G1_4 26 26 G1_9 27 27 G1_26 28 28 G2_8 29 29 G1_1 30 30 G2_30 31 31 G2_31 32 32 G1_20 33 33 G1_15 34 34 G1_29 35 35 G1_3 36 36 G1_21 37 37 G1_8 38 38 G1_13 39 39 G1 40 40 G1_12 41 41 G1_28 42 42 G1_23 43 43 G2_16 44 44 G1_17 45 45 G2_15 46 46 G1_11 47 47 G1_18 48 48 G1_10 49 49 G1_14 50 50 G2_2 51 51 S_6 52 52 G1_5 53 53 G1_24 54 54 G1_6 55 55 G2_21 56 56 G1_2 57 57 G1_22 58 58 S_17 59 59 S_7 60 60 S_9 61 61 S_14 62 62 S_23 63 63 S_15 64 64 S_1 65 65 S_30 66 66 G2_9 67 67 S_26 68 68 S_8 69 69 S_24 70 70 G1_19 71 71 G1_27 72 72 S_5 73 73 S_31 74 74 G2_26 75 75 S_27 76 76 S_4 77 77 S_25 78 78 S_11 79 79 S_13 80 80 S_19 81 81 S_16 82 82 S_29 83 83 S_22 84 84 S_10 85 85 S_21 86 86 S_3 87 87 G2_11 88 88 S 89 89 S_12 90 90 S_28 91 91 G2_3 92 92 S_18 93 93 S_20 94 94 S_2
2.2.4.1 Plotting the pseudo-time ordering in a PCA plot¶
PlotMBpca(sc,type="order")

2.2.4.2 Plotting the pseudo-time ordering in a tSNE map¶
plotOrderTsne(sc)

2.2.4.3 Plotting the Model-based clusters in heatmap¶
clustheatmap(sc,clustering_method = "model-based")
Clustering k = 1,2,..., K.max (= 20): .. done Bootstrapping, b = 1,2,..., B (= 50) [one "." per sample]: .................................................. 50
- 2
- 1
- 3

2.2.4.4 Plotting the gene expression of a particular gene in a tSNE map¶
g='ENSG00000210082' #### Plotting the expression of MT-RNR2
plotExptSNE(sc,g)

2.2.4.5 Plotting the gene expression of a particular gene in a PCA plot¶
g <- "ENSG00000210082" #### Plotting the expression of MT-RNR2
PlotMBpca(sc,g,type="exp")

g <- "ENSG00000176890" #### Plotting the expression of TYMS
PlotMBpca(sc,g,type="exp")

3. Determining differentially expressed genes (DEGs)¶
Differentially expressed genes between individual clusters are identified using the significance analysis of sequencing data (SAMseq), which is a new function in significance analysis of microarrays (Li and Tibshirani 2011) in the samr package v2.0 (Tibshirani et all., 2015). SAMseq is a non-parametric statistical function dependent on Wilcoxon rank statistic that equalizes the sizes of the library by a resampling method accounting for the various sequencing depths. The analysis is implemented over the pure raw dataset that has the unnormalized expression read counts after excluding the ERCCs. Furthermore, DEGs in each cluster comparing to all the remaining clusters are determined using binomial differential expression, which is based on binomial counting statistics.
3.1 Identifying DEGs using SAMseq¶
The user can define DEGs between all clusters generated by either K-means or model based clustering by applying the “DEGanalysis” function. Another alternative is to define DEGs between particular clusters generated by either K-means or model based clustering by applying the “DEGanalysis2clust” function. The outcome of these two functions is a list of two components:
- The first component is a data frame showing the Ensembl gage name and the symbole of the detected DEGs
- The second component is table showing the comparisons, Target cluster, Number of genes and the File name. This component will be used for the downstream analysis.
3.1.1 Determining DEGs between two particular clusters¶
####### differential expression analysis between cluster 2 and cluster 3 of the Model-Based clustering using FDR of 0.05
MBcdiff<-DEGanalysis2clust(sc,Clustering="MB",K=3,fdr=0.05,name="Name",First="CL2",Second="CL3",export = TRUE,quiet=TRUE)
#### To show the result table
head(MBcdiff[[1]]) # The first component
head(MBcdiff[[2]]) # The second component
The results of DEGs are saved in your directory Low-regulated genes in the CL3 in CL2 VS CL3 The results of DEGs are saved in your directory Up-regulated genes in the CL3 in CL2 VS CL3
| DEGsE | DEGsS |
|---|---|
| ENSG00000010292 | NCAPD2 |
| ENSG00000011426 | ANLN |
| ENSG00000024526 | DEPDC1 |
| ENSG00000072571 | HMMR |
| ENSG00000076382 | SPAG5 |
| ENSG00000079616 | KIF22 |
| Comparisons | Target cluster | Gene number | File name | Gene number | File name | |
|---|---|---|---|---|---|---|
| <chr> | <chr> | <int> | <chr> | <int> | <chr> | |
| 1 | CL2 VS CL3 | CL3 | 26 | Up-regulated-NameCL3inCL2VSCL3.csv | 146 | Low-regulated-NameCL3inCL2VSCL3.csv |
| 2 | CL2 VS CL3 | CL2 | 26 | Low-regulated-NameCL2inCL2VSCL3.csv | 146 | Up-regulated-NameCL2inCL2VSCL3.csv |

3.1.2 Determining DEGs between all clusters¶
¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤ Running this cell will overwrite the previous one ¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¶
MBcdiff<-DEGanalysis(sc,Clustering="MB",K=3,fdr=0.05,name="all_clusters",export = TRUE,quiet=TRUE) ####### differential expression analysis between all clusters
#### To show the result table
head(MBcdiff[[1]]) # The first component
head(MBcdiff[[2]]) # The second component
Estimating sequencing depths... Resampling to get new data matrices... perm = 1 perm = 2 perm = 3 perm = 4 perm = 5 perm = 6 perm = 7 perm = 8 perm = 9 perm = 10 perm = 11 perm = 12 perm = 13 perm = 14 perm = 15 perm = 16 perm = 17 perm = 18 perm = 19 perm = 20 perm = 21 perm = 22 perm = 23 perm = 24 perm = 25 perm = 26 perm = 27 perm = 28 perm = 29 perm = 30 perm = 31 perm = 32 perm = 33 perm = 34 perm = 35 perm = 36 perm = 37 perm = 38 perm = 39 perm = 40 perm = 41 perm = 42 perm = 43 perm = 44 perm = 45 perm = 46 perm = 47 perm = 48 perm = 49 perm = 50 perm = 51 perm = 52 perm = 53 perm = 54 perm = 55 perm = 56 perm = 57 perm = 58 perm = 59 perm = 60 perm = 61 perm = 62 perm = 63 perm = 64 perm = 65 perm = 66 perm = 67 perm = 68 perm = 69 perm = 70 perm = 71 perm = 72 perm = 73 perm = 74 perm = 75 perm = 76 perm = 77 perm = 78 perm = 79 perm = 80 perm = 81 perm = 82 perm = 83 perm = 84 perm = 85 perm = 86 perm = 87 perm = 88 perm = 89 perm = 90 perm = 91 perm = 92 perm = 93 perm = 94 perm = 95 perm = 96 perm = 97 perm = 98 perm = 99 perm = 100 Estimating sequencing depths... Resampling to get new data matrices... perm = 1 perm = 2 perm = 3 perm = 4 perm = 5 perm = 6 perm = 7 perm = 8 perm = 9 perm = 10 perm = 11 perm = 12 perm = 13 perm = 14 perm = 15 perm = 16 perm = 17 perm = 18 perm = 19 perm = 20 perm = 21 perm = 22 perm = 23 perm = 24 perm = 25 perm = 26 perm = 27 perm = 28 perm = 29 perm = 30 perm = 31 perm = 32 perm = 33 perm = 34 perm = 35 perm = 36 perm = 37 perm = 38 perm = 39 perm = 40 perm = 41 perm = 42 perm = 43 perm = 44 perm = 45 perm = 46 perm = 47 perm = 48 perm = 49 perm = 50 perm = 51 perm = 52 perm = 53 perm = 54 perm = 55 perm = 56 perm = 57 perm = 58 perm = 59 perm = 60 perm = 61 perm = 62 perm = 63 perm = 64 perm = 65 perm = 66 perm = 67 perm = 68 perm = 69 perm = 70 perm = 71 perm = 72 perm = 73 perm = 74 perm = 75 perm = 76 perm = 77 perm = 78 perm = 79 perm = 80 perm = 81 perm = 82 perm = 83 perm = 84 perm = 85 perm = 86 perm = 87 perm = 88 perm = 89 perm = 90 perm = 91 perm = 92 perm = 93 perm = 94 perm = 95 perm = 96 perm = 97 perm = 98 perm = 99 perm = 100 Estimating sequencing depths... Resampling to get new data matrices... perm = 1 perm = 2 perm = 3 perm = 4 perm = 5 perm = 6 perm = 7 perm = 8 perm = 9 perm = 10 perm = 11 perm = 12 perm = 13 perm = 14 perm = 15 perm = 16 perm = 17 perm = 18 perm = 19 perm = 20 perm = 21 perm = 22 perm = 23 perm = 24 perm = 25 perm = 26 perm = 27 perm = 28 perm = 29 perm = 30 perm = 31 perm = 32 perm = 33 perm = 34 perm = 35 perm = 36 perm = 37 perm = 38 perm = 39 perm = 40 perm = 41 perm = 42 perm = 43 perm = 44 perm = 45 perm = 46 perm = 47 perm = 48 perm = 49 perm = 50 perm = 51 perm = 52 perm = 53 perm = 54 perm = 55 perm = 56 perm = 57 perm = 58 perm = 59 perm = 60 perm = 61 perm = 62 perm = 63 perm = 64 perm = 65 perm = 66 perm = 67 perm = 68 perm = 69 perm = 70 perm = 71 perm = 72 perm = 73 perm = 74 perm = 75 perm = 76 perm = 77 perm = 78 perm = 79 perm = 80 perm = 81 perm = 82 perm = 83 perm = 84 perm = 85 perm = 86 perm = 87 perm = 88 perm = 89 perm = 90 perm = 91 perm = 92 perm = 93 perm = 94 perm = 95 perm = 96 perm = 97 perm = 98 perm = 99 perm = 100


| DEGsE | DEGsS |
|---|---|
| ENSG00000010292 | NCAPD2 |
| ENSG00000011426 | ANLN |
| ENSG00000024526 | DEPDC1 |
| ENSG00000072571 | HMMR |
| ENSG00000076382 | SPAG5 |
| ENSG00000079616 | KIF22 |
| Comparisons | Target cluster | Gene number | File name | Gene number | File name | |
|---|---|---|---|---|---|---|
| <chr> | <chr> | <int> | <chr> | <int> | <chr> | |
| 1 | Cl2 VS Cl3 | Cl3 | 26 | Up-regulated-all_clustersCl3inCl2VSCl3.csv | 146 | Low-regulated-all_clustersCl3inCl2VSCl3.csv |
| 2 | Cl2 VS Cl1 | Cl1 | 12 | Up-regulated-all_clustersCl1inCl2VSCl1.csv | 115 | Low-regulated-all_clustersCl1inCl2VSCl1.csv |
| 3 | Cl3 VS Cl1 | Cl1 | 0 | Up-regulated-all_clustersCl1inCl3VSCl1.csv | 10 | Low-regulated-all_clustersCl1inCl3VSCl1.csv |
| 4 | Cl2 VS Cl3 | Cl2 | 26 | Low-regulated-all_clustersCl2inCl2VSCl3.csv | 146 | Up-regulated-all_clustersCl2inCl2VSCl3.csv |
| 5 | Cl2 VS Cl1 | Cl2 | 12 | Low-regulated-all_clustersCl2inCl2VSCl1.csv | 115 | Up-regulated-all_clustersCl2inCl2VSCl1.csv |
| 6 | Cl3 VS Cl1 | Cl3 | 0 | Low-regulated-all_clustersCl3inCl3VSCl1.csv | 10 | Up-regulated-all_clustersCl3inCl3VSCl1.csv |

3.2 Identifying DEGs using binomial differential expression¶
The function MBClustDiffGenes identifies differentially regulated genes for each cluster of the Model-Based clustering in comparison to the ensemble of all cells. It returns a list with a data.frame element for each cluster that contains the mean expression across all cells not in the cluster (mean.ncl) and in the cluster (mean.cl), the fold-change in the cluster versus all remaining cells (fc), and the p-value for differential expression between all cells in a cluster and all remaining cells. The p-value is computed based on the overlap of negative binomials fitted to the count distributions within the two groups akin to DESeq.
MBcdiffBinomial<-ClustDiffGenes(sc,K=3,fdr=.01,export=TRUE, quiet=T) ########## Binomial differential expression analysis
#### To show the result table
head(MBcdiffBinomial[[1]]) # The first component
head(MBcdiffBinomial[[2]]) # The second component
| DEGsE | DEGsS |
|---|---|
| ENSG00000075624 | ACTB |
| ENSG00000094804 | CDC6 |
| ENSG00000103187 | COTL1 |
| ENSG00000112118 | MCM3 |
| ENSG00000115263 | GCG |
| ENSG00000116062 | MSH6 |
| Target Cluster | VS | Gene number | File name | Gene number | File name | |
|---|---|---|---|---|---|---|
| <chr> | <chr> | <int> | <chr> | <int> | <chr> | |
| 1 | Cluster 1 | Remaining Clusters | 9 | Up-DEG-cluster1.csv | 21 | Down-DEG-cluster1.csv |
| 2 | Cluster 2 | Remaining Clusters | 314 | Up-DEG-cluster2.csv | 295 | Down-DEG-cluster2.csv |
| 3 | Cluster 3 | Remaining Clusters | 53 | Up-DEG-cluster3.csv | 109 | Down-DEG-cluster3.csv |
Plotting the DEGs¶
Volcano plots are used to readily show the DEGs by plotting significance versus fold-change on the y and x axes, respectively.
name<-MBcdiffBinomial[[2]][2,4] ############ Selecting the "Up-DEG-cluster2.csv " from the DEGs' binomial table ##############
U<-read.csv(file=paste0(name),head=TRUE,sep=",")
Vplot<-VolcanoPlot(U,value=0.0001,name=name,FS=0.8,fc=0.75)

4. Identifying biomarkers (decision trees and networking analysis)¶
There are several methods to identify biomarkers, among them are decision trees and hub detection through networking analysis. The outcome of STRING analysis is stored in tab separated values (TSV) files. These TSV files served as an input to check both the connectivity degree and the betweenness centrality, which reflects the communication flow in the defined PPI networks
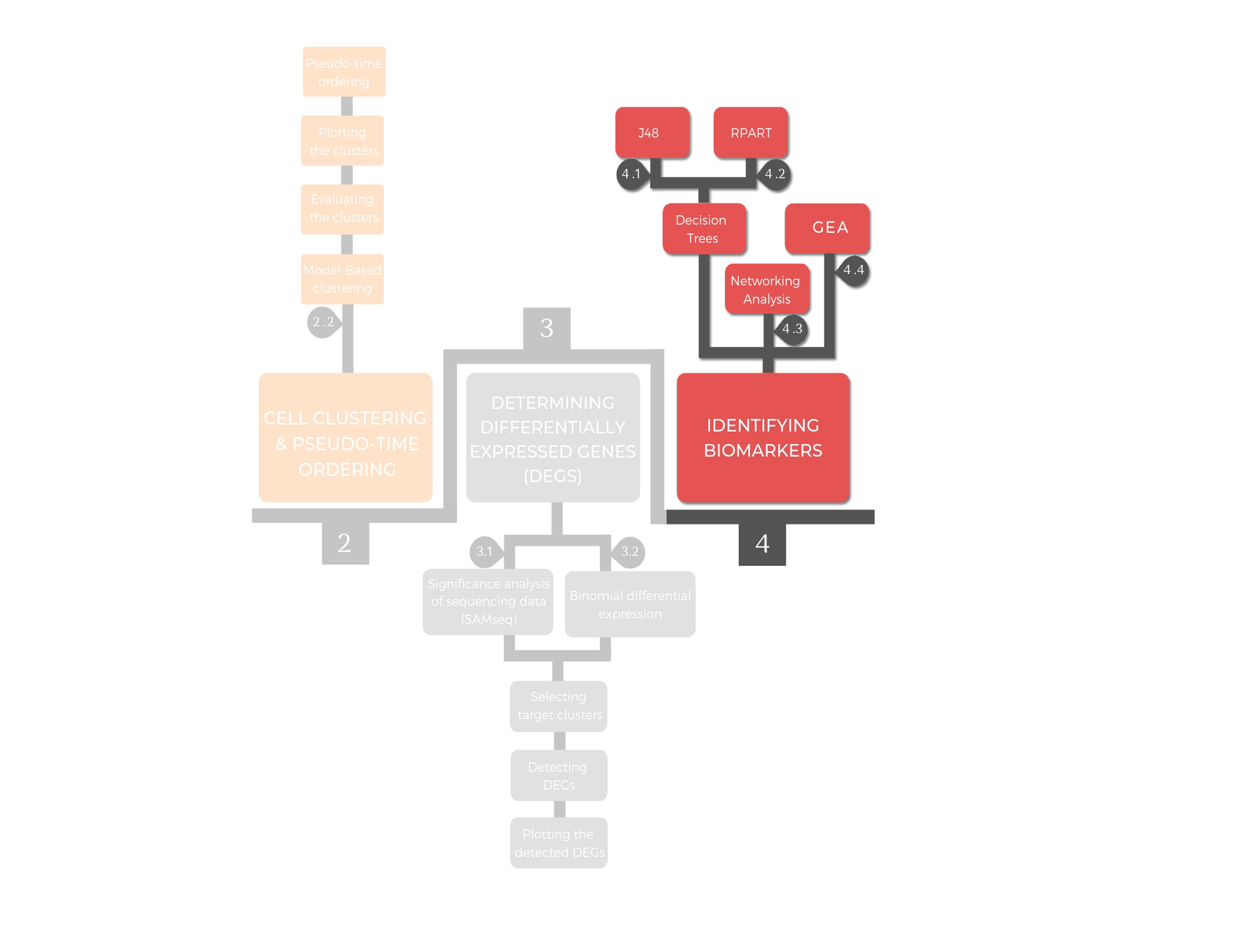
Decision trees are one of the most efficient classification techniques in biomarkers discovery. Here we use it to predict the sub-population of a target cell based on transcriptomic data. Two types of decision trees can be performed: classification and regression trees (CART) and J48. The decision tree analysis is implemented over a training dataset, which consisted of the DEGs obtained by either SAMseq or the binomial differential expression. The performance of the generated trees can be evaluated for error estimation by ten-fold cross validation assessment using the "J48DTeval" and "RpartEVAL" functions. The decision tree analysis requires the dataset to be class vectored by applying the “ClassVectoringDT” function.
sigDEG<-MBcdiff[[1]] # DEGs gene list from SANseq
#sigDEG<-MBcdiffBinomial[[1]] # DEGs gene list from Binomial analysis
First="CL2"
Second="CL3"
DATAforDT<-ClassVectoringDT(sc,Clustering="MB",K=3,First=First,Second=Second,sigDEG)
The DEGs filtered normalized dataset contains: Genes: 213 cells: 46
4.1. J48 Decision Tree¶
j48dt<-J48DT(DATAforDT)
summary(j48dt)
J48 pruned tree ------------------ KIF20A <= 471.86359: CL3 (30.0/1.0) KIF20A > 471.86359: CL2 (16.0) Number of Leaves : 2 Size of the tree : 3
=== Summary === Correctly Classified Instances 45 97.8261 % Incorrectly Classified Instances 1 2.1739 % Kappa statistic 0.9528 Mean absolute error 0.042 Root mean squared error 0.145 Relative absolute error 8.9922 % Root relative squared error 30.0308 % Total Number of Instances 46 === Confusion Matrix === a b <-- classified as 16 1 | a = CL2 0 29 | b = CL3

4.1.1. Evaluating the performance of the J48 Decision Tree¶
j48dt<-J48DTeval(DATAforDT,num.folds=10,First=First,Second=Second)
Fold 1 of 10 Fold 2 of 10 Fold 3 of 10 Fold 4 of 10 Fold 5 of 10 Fold 6 of 10 Fold 7 of 10 Fold 8 of 10 Fold 9 of 10 Fold 10 of 10
TP FN FP TN
14 3 4 25
CL2 CL3
PredictedCL2 14 4
PredictedCL3 3 25
J48 SN: 0.82 J48 SP: 0.86 J48 ACC: 0.85 J48 MCC: 0.68
4.2. RPART Decision Tree¶
rpartDT<-RpartDT(DATAforDT)
n= 46
node), split, n, loss, yval, (yprob)
* denotes terminal node
1) root 46 17 CL3 (0.36956522 0.63043478)
2) AURKA>=543.8824 18 1 CL2 (0.94444444 0.05555556)
4) CDC27>=68.20032 17 0 CL2 (1.00000000 0.00000000) *
5) CDC27< 68.20032 1 0 CL3 (0.00000000 1.00000000) *
3) AURKA< 543.8824 28 0 CL3 (0.00000000 1.00000000) *

4.2.1. Evaluating the performance of the RPART Decision Tree¶
rpartEVAL<-RpartEVAL(DATAforDT,num.folds=10,First=First,Second=Second)
Fold 1 of 10 Fold 2 of 10 Fold 3 of 10 Fold 4 of 10 Fold 5 of 10 Fold 6 of 10 Fold 7 of 10 Fold 8 of 10 Fold 9 of 10 Fold 10 of 10
TP FN FP TN
12 5 3 26
CL2 CL3
PredictedCL2 12 3
PredictedCL3 5 26
Rpart SN: 0.71 Rpart SP: 0.9 Rpart ACC: 0.83 Rpart MCC: 0.62
4.3. Networking Analysis¶
To define protein-protein interactions (PPI) over a list of genes, STRING-api is used. The outcome of STRING analysis was stored in tab separated values (TSV) files. These TSV files served as an input to check both the connectivity degree and the betweenness centrality, which reflects the communication flow in the defined PPI networks.
4.3.1 All DEGs¶
DEGs="All DEGs"
FileName=paste0(DEGs)
#data<-MBcdiffBinomial[[1]] [,2] # DEGs gene list from Binomial analysis
data<-MBcdiff[[1]] [,2] # From the table of the differential expression analysis between all pairs of clusters
ppi<-PPI(data,FileName)
networking<-NetAnalysis(ppi)
networking ##### In case the Examine response components = 200 and an error "linkmat[i, ]" appeared, that means there are no PPI.
# Plotting the network of the top 25 DEGs
DATA<-data[1:25]
network<-Networking(DATA,FileName,plot_width = 25, plot_height = 10)
Retrieving URL. Please wait... Successful retrieval. ── Column specification ──────────────────────────────────────────────────────── cols( stringId_A = col_character(), stringId_B = col_character(), preferredName_A = col_character(), preferredName_B = col_character(), ncbiTaxonId = col_double(), score = col_double(), nscore = col_double(), fscore = col_double(), pscore = col_double(), ascore = col_double(), escore = col_double(), dscore = col_double(), tscore = col_double() ) Number of nodes: 168 Number of links: 3909 Link Density: 23.2678571428571 The connectance of the graph: 0.139328485885372 Mean Distences1.67691387559809 Average Path Length1.67691387559809
| names | degree | betweenness | |
|---|---|---|---|
| <chr> | <dbl> | <dbl> | |
| 110 | CDC20 | 108 | 200.85262 |
| 56 | PLK1 | 106 | 161.50984 |
| 127 | CDK1 | 106 | 205.26652 |
| 23 | CCNB1 | 103 | 46.45923 |
| 53 | MAD2L1 | 102 | 91.57888 |
| 29 | NDC80 | 100 | 60.54970 |
| 46 | CCNA2 | 100 | 158.46909 |
| 49 | BUB1B | 100 | 94.02535 |
| 50 | CCNB2 | 100 | 41.30413 |
| 62 | BUB1 | 100 | 153.89862 |
| 69 | AURKB | 100 | 160.84723 |
| 28 | KIF11 | 99 | 65.62777 |
| 108 | KIF2C | 99 | 131.15731 |
| 7 | AURKA | 98 | 13.08012 |
| 111 | CDCA8 | 97 | 102.68655 |
| 136 | TOP2A | 96 | 40.91966 |
| 39 | CENPE | 95 | 38.29335 |
| 97 | CENPF | 95 | 57.51430 |
| 44 | NUF2 | 94 | 56.21489 |
| 61 | BIRC5 | 94 | 66.33167 |
| 103 | TTK | 94 | 84.73181 |
| 58 | TPX2 | 93 | 104.19420 |
| 90 | UBE2C | 93 | 238.76019 |
| 124 | PRC1 | 92 | 59.66206 |
| 16 | DLGAP5 | 90 | 27.89618 |
| 55 | MELK | 90 | 34.65601 |
| 100 | ASPM | 89 | 50.66924 |
| 125 | KIF20A | 89 | 45.00967 |
| 27 | KIF23 | 88 | 14.97698 |
| 122 | PTTG1 | 88 | 66.48604 |
| ⋮ | ⋮ | ⋮ | ⋮ |
| 107 | IFIT1 | 2 | 1 |
| 112 | SLC25A25 | 2 | 0 |
| 130 | CALCOCO2 | 2 | 86 |
| 157 | LDHB | 2 | 0 |
| 5 | MTHFD1 | 1 | 0 |
| 10 | SLC25A3 | 1 | 0 |
| 25 | MRPL15 | 1 | 0 |
| 32 | COTL1 | 1 | 0 |
| 43 | TIMP3 | 1 | 0 |
| 57 | CLPX | 1 | 0 |
| 82 | PTTG1IP | 1 | 0 |
| 83 | PHF7 | 1 | 0 |
| 86 | CMTR2 | 1 | 0 |
| 87 | PTK2 | 1 | 0 |
| 89 | MFSD12 | 1 | 0 |
| 91 | MOB3A | 1 | 0 |
| 120 | FBXO8 | 1 | 0 |
| 126 | PSAP | 1 | 0 |
| 152 | WDR6 | 1 | 0 |
| 158 | CBR3 | 1 | 0 |
| 159 | HN1 | 1 | 0 |
| 160 | COL3A1 | 1 | 0 |
| 161 | CABYR | 1 | 0 |
| 162 | CMTR1 | 1 | 0 |
| 163 | LAMB2 | 1 | 0 |
| 164 | TMUB2 | 1 | 0 |
| 165 | RBMX | 1 | 0 |
| 166 | MXD3 | 1 | 0 |
| 167 | PHACTR4 | 1 | 0 |
| 168 | PHF19 | 1 | 0 |
Retrieving URL. Please wait... Successful retrieval. You can see the network with high resolution by clicking on the following link: https://string-db.org/api/highres_image/network?identifiers=NCAPD2%0dANLN%0dDEPDC1%0dHMMR%0dSPAG5%0dKIF22%0dNDC80%0dAURKA%0dTPX2%0dBIRC5%0dRANGAP1%0dCDKN3%0dCDC25B%0dFAM83D%0dPLIN3%0dISYNA1%0dTTK%0dKIF20A%0dSMC4%0dMIIP%0dCDC20%0dNEK2%0dCENPF%0dPHF19%0dKIF18A&species=9606

4.3.2 Particular set of DEGs¶
DEGs="Down-Reg-CL2"
FileName=paste0(DEGs)
## Networking analysis of Down-regulated genes in cluster2
name<-MBcdiff[[2]][4,4] # From the table of the differential expression analysis between all pairs of clusters
U<-read.csv(file=paste0(name),head=TRUE,sep=",")
data<- U[,3] # Selecting the gene names
ppi<-PPI(data,FileName)
networking<-NetAnalysis(ppi)
networking ##### In case the Examine response components = 200 and an error "linkmat[i, ]" appeared, that means there are no PPI.
# Plotting the network
network<-Networking(data,FileName,plot_width = 25, plot_height = 25)
Retrieving URL. Please wait... Successful retrieval. ── Column specification ──────────────────────────────────────────────────────── cols( stringId_A = col_character(), stringId_B = col_character(), preferredName_A = col_character(), preferredName_B = col_character(), ncbiTaxonId = col_double(), score = col_double(), nscore = col_double(), fscore = col_double(), pscore = col_double(), ascore = col_double(), escore = col_double(), dscore = col_double(), tscore = col_double() ) Number of nodes: 23 Number of links: 147 Link Density: 6.39130434782609 The connectance of the graph: 0.290513833992095 Mean Distences1.15789473684211 Average Path Length1.15789473684211
| names | degree | betweenness | |
|---|---|---|---|
| <chr> | <dbl> | <dbl> | |
| 15 | TYMS | 18 | 7.7107143 |
| 19 | RFC4 | 18 | 8.0107143 |
| 2 | MCM5 | 17 | 1.0000000 |
| 9 | MCM4 | 17 | 1.7107143 |
| 12 | MCM2 | 17 | 0.7107143 |
| 13 | FEN1 | 17 | 0.7107143 |
| 14 | MCM7 | 17 | 0.7107143 |
| 17 | PCNA | 17 | 0.5107143 |
| 1 | CDC6 | 16 | 0.0000000 |
| 7 | GINS2 | 16 | 0.1000000 |
| 11 | MCM6 | 16 | 0.1000000 |
| 16 | RRM2 | 16 | 0.1000000 |
| 21 | CDC45 | 16 | 0.5000000 |
| 6 | GMNN | 15 | 0.0000000 |
| 18 | DUT | 15 | 0.1250000 |
| 22 | CCNE2 | 15 | 0.0000000 |
| 20 | HELLS | 14 | 0.0000000 |
| 4 | APEX1 | 9 | 0.0000000 |
| 10 | RPL8 | 3 | 5.0000000 |
| 23 | LDHB | 2 | 0.0000000 |
| 3 | MTHFD1 | 1 | 0.0000000 |
| 5 | SLC25A3 | 1 | 0.0000000 |
| 8 | MRPL15 | 1 | 0.0000000 |
Retrieving URL. Please wait... Successful retrieval. You can see the network with high resolution by clicking on the following link: https://string-db.org/api/highres_image/network?identifiers=MCM6%0dMCM5%0dMCM4%0dPCNA%0dFEN1%0dCCNE2%0dTYMS%0dH4C3%0dCDC45%0dMTHFD1%0dLDHB%0dDUT%0dMCM7%0dC19orf48%0dMCM2%0dC1QBP%0dGINS2%0dSLC25A3%0dCDC6%0dRPL8%0dAPEX1%0dHELLS%0dMRPL15%0dRFC4%0dRRM2%0dGMNN&species=9606

4.4 Gene Enrichment Analysis¶
dbs <- listEnrichrDbs()
head(dbs)
#print(dbs)
| geneCoverage | genesPerTerm | libraryName | link | numTerms | |
|---|---|---|---|---|---|
| <dbl> | <dbl> | <chr> | <chr> | <dbl> | |
| 1 | 13362 | 275 | Genome_Browser_PWMs | http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/ | 615 |
| 2 | 27884 | 1284 | TRANSFAC_and_JASPAR_PWMs | http://jaspar.genereg.net/html/DOWNLOAD/ | 326 |
| 3 | 6002 | 77 | Transcription_Factor_PPIs | 290 | |
| 4 | 47172 | 1370 | ChEA_2013 | http://amp.pharm.mssm.edu/lib/cheadownload.jsp | 353 |
| 5 | 47107 | 509 | Drug_Perturbations_from_GEO_2014 | http://www.ncbi.nlm.nih.gov/geo/ | 701 |
| 6 | 21493 | 3713 | ENCODE_TF_ChIP-seq_2014 | http://genome.ucsc.edu/ENCODE/downloads.html | 498 |
############ Up-regulated genes in cluster 2 ##############
#DEGs=MBcdiffBinomial[[2]][2,4] # Up-regulated genes in cluster 2 (from the Binomial analysis)
DEGs=MBcdiff[[2]][2,6] # UP-regulated genes in cluster 2 (from SAMseq)
data<-read.csv(file=paste0(DEGs),head=TRUE,sep=",")
data<-as.character(data[,3])
dbs <- c("KEGG_2019_Human","GO_Biological_Process_2018")
enriched <- enrichr(data, dbs)
KEGG_2019_Human<-enriched[[1]][,c(1,2,3,9)]
GO_Biological_Process_2018<-enriched[[2]][,c(1,2,3,9)]
GEA<-rbind(KEGG_2019_Human,GO_Biological_Process_2018)
GEA
Uploading data to Enrichr... Done. Querying KEGG_2019_Human... Done. Querying GO_Biological_Process_2018... Done. Parsing results... Done.
| Term | Overlap | P.value | Genes |
|---|---|---|---|
| <chr> | <chr> | <dbl> | <chr> |
| Cell cycle | 13/124 | 3.015997e-13 | PLK1;BUB1B;TTK;CDC25B;CDC20;CCNA2;CCNB2;CCNB1;CDC27;CDK1;BUB3;BUB1;MAD2L1 |
| Oocyte meiosis | 12/125 | 7.430102e-12 | CDC20;CCNB2;CCNB1;CDC27;PLK1;CDK1;PPP2R5D;FBXO43;CALM2;BUB1;AURKA;MAD2L1 |
| Progesterone-mediated oocyte maturation | 11/99 | 1.150315e-11 | CCNA2;CCNB2;CCNB1;CDC27;PLK1;CDK1;KIF22;BUB1;CDC25B;AURKA;MAD2L1 |
| Gap junction | 5/88 | 1.542720e-04 | TUBA1C;TUBB6;TUBB2A;CDK1;TUBB4B |
| Human T-cell leukemia virus 1 infection | 7/219 | 2.762843e-04 | CDC20;CCNA2;CCNB2;CDC27;BUB1B;BUB3;MAD2L1 |
| Pathogenic Escherichia coli infection | 4/55 | 2.821654e-04 | TUBA1C;TUBB6;TUBB2A;TUBB4B |
| Phagosome | 5/152 | 1.862814e-03 | TUBA1C;TUBB6;TUBB2A;ATP6V1B2;TUBB4B |
| Cellular senescence | 5/160 | 2.329622e-03 | CCNA2;CCNB2;CCNB1;CDK1;CALM2 |
| p53 signaling pathway | 3/72 | 8.281581e-03 | CCNB2;CCNB1;CDK1 |
| Human immunodeficiency virus 1 infection | 4/212 | 3.414105e-02 | CCNB2;CCNB1;CDK1;CALM2 |
| FoxO signaling pathway | 3/132 | 4.067356e-02 | CCNB2;CCNB1;PLK1 |
| Ubiquitin mediated proteolysis | 3/137 | 4.459546e-02 | CDC20;UBE2C;CDC27 |
| RNA degradation | 2/79 | 7.589034e-02 | TOB1;DCP2 |
| Small cell lung cancer | 2/93 | 1.001516e-01 | CKS2;CKS1B |
| Viral carcinogenesis | 3/201 | 1.096414e-01 | CDC20;CCNA2;CDK1 |
| Vitamin digestion and absorption | 1/24 | 1.293174e-01 | BTD |
| Maturity onset diabetes of the young | 1/26 | 1.393136e-01 | PAX6 |
| Collecting duct acid secretion | 1/27 | 1.442690e-01 | ATP6V1B2 |
| Phototransduction | 1/28 | 1.491961e-01 | CALM2 |
| AMPK signaling pathway | 2/120 | 1.516936e-01 | CCNA2;PPP2R5D |
| Dopaminergic synapse | 2/131 | 1.739591e-01 | PPP2R5D;CALM2 |
| Apoptosis | 2/143 | 1.987992e-01 | TUBA1C;BIRC5 |
| Adrenergic signaling in cardiomyocytes | 2/145 | 2.029824e-01 | PPP2R5D;CALM2 |
| Hepatitis B | 2/163 | 2.409985e-01 | CCNA2;BIRC5 |
| Amino sugar and nucleotide sugar metabolism | 1/48 | 2.420385e-01 | CYB5RL |
| Vibrio cholerae infection | 1/50 | 2.507510e-01 | ATP6V1B2 |
| Cholesterol metabolism | 1/50 | 2.507510e-01 | LDLRAP1 |
| Human papillomavirus infection | 3/330 | 2.952720e-01 | CCNA2;ATP6V1B2;PPP2R5D |
| Arachidonic acid metabolism | 1/63 | 3.050120e-01 | CBR3 |
| Mitophagy | 1/65 | 3.130066e-01 | CALCOCO2 |
| ⋮ | ⋮ | ⋮ | ⋮ |
| regulation of MAPK cascade (GO:0043408) | 1/203 | 0.6916701 | FAM83D |
| positive regulation of signal transduction (GO:0009967) | 1/206 | 0.6970124 | LANCL2 |
| regulation of translation (GO:0006417) | 1/213 | 0.7091236 | TOB1 |
| protein oligomerization (GO:0051259) | 1/217 | 0.7158275 | RBMX |
| ion transmembrane transport (GO:0034220) | 1/219 | 0.7191218 | ATP6V1B2 |
| viral process (GO:0016032) | 1/220 | 0.7207548 | CALCOCO2 |
| endosomal transport (GO:0016197) | 1/229 | 0.7350339 | VPS26B |
| regulation of signal transduction (GO:0009966) | 1/233 | 0.7411456 | BIRC5 |
| RNA splicing, via transesterification reactions with bulged adenosine as nucleophile (GO:0000377) | 1/236 | 0.7456374 | RBMX |
| plasma membrane bounded cell projection assembly (GO:0120031) | 1/241 | 0.7529527 | HYLS1 |
| defense response to bacterium (GO:0042742) | 1/241 | 0.7529527 | HMGB2 |
| regulation of ERK1 and ERK2 cascade (GO:0070372) | 1/247 | 0.7614563 | FAM83D |
| mRNA splicing, via spliceosome (GO:0000398) | 1/261 | 0.7801861 | RBMX |
| endocytosis (GO:0006897) | 1/263 | 0.7827400 | LDLRAP1 |
| regulation of programmed cell death (GO:0043067) | 1/268 | 0.7889967 | BIRC5 |
| cellular response to oxygen-containing compound (GO:1901701) | 1/274 | 0.7962693 | HMGB2 |
| mRNA processing (GO:0006397) | 1/283 | 0.8067149 | RBMX |
| negative regulation of signal transduction (GO:0009968) | 1/283 | 0.8067149 | PHACTR4 |
| ion transport (GO:0006811) | 1/289 | 0.8133819 | ATP6V1B2 |
| proteolysis (GO:0006508) | 1/291 | 0.8155531 | CLPX |
| cellular protein localization (GO:0034613) | 1/327 | 0.8506184 | VPS26B |
| intracellular protein transport (GO:0006886) | 1/347 | 0.8671535 | VPS26B |
| post-translational protein modification (GO:0043687) | 1/357 | 0.8747271 | CCNF |
| cytokine-mediated signaling pathway (GO:0019221) | 2/633 | 0.8828956 | BIRC5;IFIT1 |
| transmembrane transport (GO:0055085) | 1/378 | 0.8892664 | ATP6V1B2 |
| transmembrane receptor protein tyrosine kinase signaling pathway (GO:0007169) | 1/396 | 0.9003883 | ATP6V1B2 |
| gene expression (GO:0010467) | 1/411 | 0.9088052 | CALCOCO1 |
| neutrophil degranulation (GO:0043312) | 1/479 | 0.9389349 | TUBB4B |
| neutrophil activation involved in immune response (GO:0002283) | 1/483 | 0.9403613 | TUBB4B |
| neutrophil mediated immunity (GO:0002446) | 1/487 | 0.9417546 | TUBB4B |
############ Up-regulated genes in cluster 2 ##############
DEGs=MBcdiffBinomial[[2]][2,4] # Up-regulated genes in cluster 2 (from the Binomial analysis)
data<-read.csv(file=paste0(DEGs),head=TRUE,sep=",")
data<-as.character(data[,3])
dbs <- c("KEGG_2019_Human","GO_Biological_Process_2018")
enriched <- enrichr(data, dbs)
KEGG_2019_Human<-enriched[[1]][,c(1,2,3,9)]
GO_Biological_Process_2018<-enriched[[2]][,c(1,2,3,9)]
GEA<-rbind(KEGG_2019_Human,GO_Biological_Process_2018)
GEA
Uploading data to Enrichr... Done. Querying KEGG_2019_Human... Done. Querying GO_Biological_Process_2018... Done. Parsing results... Done.
| Term | Overlap | P.value | Genes |
|---|---|---|---|
| <chr> | <chr> | <dbl> | <chr> |
| Cell cycle | 19/124 | 7.918476e-14 | PLK1;BUB1B;TTK;CDC25B;CDC20;CCNA2;RBL2;MAD2L2;CCNB2;CCNB1;DBF4;PTTG1;RAD21;CDC27;CDK1;ANAPC5;BUB3;BUB1;MAD2L1 |
| Oocyte meiosis | 17/125 | 1.202700e-11 | PLK1;PPP2R5D;PPP2R5C;FBXO43;AURKA;CDC20;SGO1;MAD2L2;CCNB2;CCNB1;PTTG1;CDC27;CDK1;ANAPC5;CALM2;BUB1;MAD2L1 |
| Progesterone-mediated oocyte maturation | 13/99 | 5.070882e-09 | PLK1;KIF22;CDC25B;AURKA;CCNA2;MAD2L2;CCNB2;CCNB1;CDC27;CDK1;ANAPC5;BUB1;MAD2L1 |
| Pathogenic Escherichia coli infection | 7/55 | 2.339053e-05 | TUBA1C;TUBB6;TUBA1A;TUBB2A;ARPC5;TUBB4B;ACTB |
| Human T-cell leukemia virus 1 infection | 11/219 | 7.194567e-04 | CDC20;FOSL1;CCNA2;CCNB2;PTTG1;CDC27;NFATC3;BUB1B;ANAPC5;BUB3;MAD2L1 |
| p53 signaling pathway | 6/72 | 9.349293e-04 | CCNB2;CCNB1;APAF1;CDK1;PMAIP1;GTSE1 |
| Cellular senescence | 9/160 | 9.827814e-04 | CCNA2;RBL2;CCNB2;CCNB1;CXCL8;NFATC3;CDK1;CALM2;ZFP36L1 |
| Apoptosis | 8/143 | 1.923972e-03 | TUBA1C;TUBA1A;APAF1;PMAIP1;BIRC5;RIPK1;SPTAN1;ACTB |
| Gap junction | 6/88 | 2.632569e-03 | TUBA1C;TUBB6;TUBA1A;TUBB2A;CDK1;TUBB4B |
| Phagosome | 7/152 | 1.029573e-02 | TUBA1C;TUBB6;TUBA1A;TUBB2A;ATP6V1B2;TUBB4B;ACTB |
| Fructose and mannose metabolism | 3/33 | 1.476794e-02 | MPI;ALDOA;HK1 |
| Small cell lung cancer | 5/93 | 1.563301e-02 | APAF1;LAMB2;CKS2;PTK2;CKS1B |
| FoxO signaling pathway | 6/132 | 1.802410e-02 | RBL2;CCNB2;CCNB1;PLK2;PLK1;SGK1 |
| Shigellosis | 4/65 | 1.907655e-02 | MAD2L2;CXCL8;ARPC5;ACTB |
| Ubiquitin mediated proteolysis | 6/137 | 2.122235e-02 | CDC20;HERC4;UBE2C;CDC27;ANAPC5;UBE2A |
| Bacterial invasion of epithelial cells | 4/74 | 2.908954e-02 | MAD2L2;ARPC5;ACTB;PTK2 |
| Cardiac muscle contraction | 4/78 | 3.437236e-02 | TPM4;COX3;COX1;CYTB |
| Amino sugar and nucleotide sugar metabolism | 3/48 | 3.946386e-02 | UGDH;MPI;HK1 |
| Viral carcinogenesis | 7/201 | 3.984129e-02 | CDC20;CCNA2;RBL2;CDK1;PMAIP1;ACTN4;GTF2E1 |
| Human immunodeficiency virus 1 infection | 7/212 | 5.048456e-02 | CCNB2;CCNB1;NFATC3;CDK1;RIPK1;CALM2;PTK2 |
| Alzheimer disease | 6/171 | 5.327641e-02 | PSENEN;APAF1;COX3;COX1;CYTB;CALM2 |
| IL-17 signaling pathway | 4/93 | 5.883211e-02 | FOSL1;CXCL8;ANAPC5;IL17RC |
| Oxidative phosphorylation | 5/133 | 5.884214e-02 | COX3;ATP6V1B2;COX1;CYTB;ND2 |
| Amoebiasis | 4/96 | 6.459788e-02 | CXCL8;LAMB2;ACTN4;PTK2 |
| Phosphatidylinositol signaling system | 4/99 | 7.064810e-02 | IMPA1;INPP5J;CALM2;MTMR6 |
| Parkinson disease | 5/142 | 7.346047e-02 | APAF1;COX3;COX1;CYTB;ND2 |
| Neomycin, kanamycin and gentamicin biosynthesis | 1/5 | 7.608058e-02 | HK1 |
| Mitophagy | 3/65 | 8.234366e-02 | USP8;CALCOCO2;CSNK2A2 |
| Galactose metabolism | 2/31 | 8.484336e-02 | GLB1;HK1 |
| Adherens junction | 3/72 | 1.040215e-01 | CSNK2A2;ACTN4;ACTB |
| ⋮ | ⋮ | ⋮ | ⋮ |
| cellular response to DNA damage stimulus (GO:0006974) | 3/329 | 0.8924690 | TOP2A;CDK1;UBE2A |
| positive regulation of macromolecule biosynthetic process (GO:0010557) | 1/142 | 0.8951364 | DBF4 |
| cell-cell adhesion via plasma-membrane adhesion molecules (GO:0098742) | 1/142 | 0.8951364 | DSG2 |
| neuron development (GO:0048666) | 1/144 | 0.8984265 | STMN1 |
| negative regulation of cell differentiation (GO:0045596) | 1/150 | 0.9076919 | MAD2L2 |
| inflammatory response (GO:0006954) | 2/252 | 0.9083321 | CXCL8;NFATC3 |
| protein stabilization (GO:0050821) | 1/152 | 0.9105892 | GTSE1 |
| ribonucleoprotein complex assembly (GO:0022618) | 1/156 | 0.9161147 | GEMIN2 |
| regulation of protein phosphorylation (GO:0001932) | 2/261 | 0.9183423 | RIPK1;PTK2 |
| positive regulation of transcription from RNA polymerase II promoter (GO:0045944) | 9/848 | 0.9199766 | OSR2;KLF6;NFATC3;HMGB2;CKAP2;PAX6;RIPK1;RBMX;DLGAP5 |
| positive regulation of cellular biosynthetic process (GO:0031328) | 1/159 | 0.9200341 | DBF4 |
| endocytosis (GO:0006897) | 2/263 | 0.9204231 | TOM1;LDLRAP1 |
| T cell receptor signaling pathway (GO:0050852) | 1/163 | 0.9249776 | BTN2A1 |
| regulation of inflammatory response (GO:0050727) | 1/166 | 0.9284842 | TMSB4X |
| nuclear-transcribed mRNA catabolic process (GO:0000956) | 1/174 | 0.9370566 | ZFP36L1 |
| transmembrane transport (GO:0055085) | 3/378 | 0.9381998 | FKBP1B;ATP6V1B2;SGK1 |
| positive regulation of MAP kinase activity (GO:0043406) | 1/177 | 0.9400002 | CDK1 |
| Fc-epsilon receptor signaling pathway (GO:0038095) | 1/182 | 0.9446043 | NFATC3 |
| Fc receptor signaling pathway (GO:0038093) | 1/183 | 0.9454820 | NFATC3 |
| positive regulation of GTPase activity (GO:0043547) | 1/192 | 0.9527827 | RANGAP1 |
| positive regulation of cell differentiation (GO:0045597) | 1/194 | 0.9542678 | ZFP36L1 |
| positive regulation of sequence-specific DNA binding transcription factor activity (GO:0051091) | 1/215 | 0.9673105 | RIPK1 |
| positive regulation of cytokine production (GO:0001819) | 1/220 | 0.9698235 | RIPK1 |
| extracellular matrix organization (GO:0030198) | 1/229 | 0.9738715 | LAMB2 |
| anterograde trans-synaptic signaling (GO:0098916) | 1/240 | 0.9780910 | UNC13B |
| defense response to bacterium (GO:0042742) | 1/241 | 0.9784391 | HMGB2 |
| regulation of ERK1 and ERK2 cascade (GO:0070372) | 1/247 | 0.9804150 | FAM83D |
| regulation of immune response (GO:0050776) | 1/251 | 0.9816308 | BTN2A1 |
| antigen receptor-mediated signaling pathway (GO:0050851) | 1/257 | 0.9833150 | BTN2A1 |
| chemical synaptic transmission (GO:0007268) | 1/289 | 0.9900141 | UNC13B |